ЮВЕНІЛЬНА СКЛЕРОДЕРМІЯ — ПРОБЛЕМНІ АСПЕКТИ
Шевченко Н.С.1, Білявська Ю.В.2, Ошлянська О.А.3
- 1ДУ «Інститут охорони здоров’я дітей та підлітків НАМН України»
- 2ДУ «ННЦ «Інститут кардіології імені акад. М.Д. Стражеска» НАМН України
- 3Національний університет охорони здоров’я України імені П.Л. Шупика
Резюме. Обґрунтування. Ювенільна склеродермія з двома її різновидами, ювенільною локалізованою склеродермією та системним склерозом, є третім за частотою ревматичним захворюванням у дитячому віці. В останні роки відбувся якісний перегляд підходів щодо діагностики, терапії та спостереження дітей із склеродермічною хворобою, які базуються на найкращих наявних доказах і містять рекомендації щодо оцінки та лікування пацієнтів з метою покращання результатів. Методи дослідження. Проаналізовано наукові дані щодо сучасних уявлень і змін поглядів на окремі позиції етіології та патогенезу, діагностичні критерії, клінічні прояви, класифікаційні параметри склеродермії в порівнянні у педіатричній та дорослій ревматології. Результати дослідження. Проаналізовано сучасний стан проблеми, зосереджено увагу не лише на оцінці стану шкіри, а й на ранніх ознаках змін внутрішніх органів, правильне лікування яких може істотно вплинути на віддалений результат. Представлено педіатричну оцінку тяжкості захворювання за багатовимірною шкалою тяжкості під назвою «J4S», яка включає параметри росту, шкіри та ураження внутрішніх органів. Визначено поетапний підхід до лікування з метою уніфікації стандартів медичної допомоги, що полегшує прийняття рішень у повсякденній практиці. Відокремлено зміни в менеджменті локалізованої склеродермії, де більш широко застосовуються клінічні та інструментальні системи оцінки. Показано консенсусне твердження на підставі довгострокових тривалих досліджень про важливу роль метотрексату в лікуванні. Висновок. Імплементація останніх рекомендацій щодо діагностики та лікування локалізованої і системної форми склеродермії у дітей необхідна для підвищення ефективності і правильного лікування, яке може істотно вплинути на віддалений результат, зниження захворюваності та смертності від цієї хвороби. Важливою залишається співпраця дорослих і дитячих ревматологів на засадах доказової медицини для зберігання стратегії лікування і подальшого спостереження хворих дітей.
DOI: 10.32471/rheumatology.2707-6970.88.16997
УДК 616-004.-616.1/.9- 616.6-07
Вступ
Відповідно до сучасних поглядів «склеродермія» — це група патологічних станів, які поєднують захворювання, загальною рисою яких є специфічне ураження шкіри. Незважаючи на увагу та інтерес до проблеми склеродермії, досить велику кількість публікацій щодо клінічних проявів та особливостей її лікування, зберігається певна частка діагностичних помилок, що насамперед стосується ранніх етапів розвитку хвороби. Дискутабельними залишаються і питання класифікації склеродермічних уражень, які раніше включали обмежену або вогнищеву та системну форми склеродермії, а зараз розділяють варіанти локалізованого і системного склерозу. У коло станів, що найбільш часто диференціюються, включається широкий спектр захворювань, у тому числі дерматологічного профілю, що переважно належали до обмеженої склеродермії, і були суто дерматологічною проблемою, а сьогодні розглядаються в колі ревматичних захворювань. Відповідно, існує широкий спектр лікарських препаратів і засобів, що застосовуються в терапії даної патології, і не завжди їх застосування відповідає міжнародним стандартам і засадам доказової медицини.
Визначення і класифікаційні підходи
Термін «склеродермія» походить від грецьких слів skleros (твердий або ущільнений) і derma (шкіра) і використовується для опису захворювання, що характеризується прогресуючим затвердінням і ущільненням шкіри. Склеродермія — це аспект системного склерозу, одного з системних захворювань сполучної тканини, яке уражує підшкірну клітковину, м’язи та внутрішні органи.
Термін «системний склероз» використовується для опису системного аутоімунного захворювання невідомого походження, що характеризується надмірним відкладенням колагену та інших макромолекул сполучної тканини в шкірі та багатьох внутрішніх органах, помітними і часто тяжкими фібропроліферативними змінами в мікроциркуляторному руслі та численними гуморальними та клітинними імунологічними аномаліями. Системний склероз найбільш очевидний у шкірі, проте часто залучаються шлунково-кишковий тракт, а також дихальна, ниркова, серцево-судинна, кістково-м’язова, ендокринна та сечостатева системи.
За визначенням Європейського альянсу асоціацій ревматологів (European Alliance of Associations for Rheumatology — EULAR) та Американського коледжу ревматологів (American College of Rheumatolo gy — ACR), системний склероз — це складне і гетерогенне захворювання з клінічними формами, що варіюють від обмеженого ураження шкіри (обмежений системний склероз шкіри) до форм з поширеним склерозом шкіри та тяжким, часто прогресуючим ураженням внутрішніх органів (дифузний системний склероз шкіри), а іноді й фульмінантним перебігом (фульмінантний системний склероз) [1, 2]. За міжнародною класифікацією хвороб ХІ перегляду, системний склероз (шифр 4A42) — це розповсюджене ураження сполучної тканини, що проявляється затвердінням і потовщенням шкіри, порушенням мікроциркуляції та більших судин, а також фіброзними дегенеративними змінами в різних органах тіла, включаючи серце, легені, нирки та шлунково-кишковий тракт [Foundation URI: http://id.who.int/icd/entity/1084365812]. Існують поодинокі випадки типового системного склерозу — ураження внутрішніх органів за відсутності клінічно очевидного ураження шкіри, клінічна підгрупа, відома як «scleroderma sine scleroderma».
Дифузний системний склероз шкіри (шифр 4A42.1) — це підтип системного склерозу, що характеризується фіброзом тулубної шкіри або ділянок кінцівок з раннім і значним поширенням дифузного ураження органів (інтерстиціальна хвороба легень, олігурична ниркова недостатність, дифузне захворювання шлунково-кишкового тракту та ураження міокарда).
Обмежений системний склероз (шифр 4A42.2), за сучасним визначенням, включає поєднання кальцинозу, феномену Рейно, дисфункції стравоходу, склеродактилії та телеангіектазії, а також наявність антитіл до топоізомерази I (анти-Scl70) і РНК-полімерази III (анти-RNAP III). Як правило, обмежений системний склероз шкіри охоплює ділянки дистальніше ліктів і колін, але може уражати обличчя і шию. Раніше ця форма відповідала синдрому CREST, давньому терміну, який використовувався для опису цієї підгрупи обмеженого шкірного системного склерозу, хоча не всі необхідні складові є для того, щоб розлад відповідав класичному CREST.
Окремо визначається і системний склероз у дітей (шифр 4A42.0) — захворювання, що виникає у віці до 16 років, при якому ураження внутрішніх органів відмічається рідше, але артрит і міозит — частіше, ніж у дорослих [Foundation URI : http://id.who.int/icd/entity/2338375]. На відміну від дорослих, в педіатричній практиці відокремлюють ювенільну системну склеродермію (ССД) з дифузним ураженням шкіри (дифузна форма) або з лімітованим ураженням шкіри. Також мають місце перехресні форми — «overlap»-синдроми [3, 4]. Ювенільна склеродермія в дитячому віці виявляється частіше, є третім за частотою хронічним ревматичним захворюванням в педіатричній популяції після ювенільного ідіопатичного артриту і системного червоного вовчака [4, 5, 6].
Форми, які не відповідають описаним чітким характеристикам, об’єднуються під шифром 4A42.Z — системний склероз неуточнений. Слід зазначити, що при недиференційованих варіантах системних захворювань сполучної тканини у третини дітей відмічають старт системного склерозу, як і можливість розвитку інших ревматичних захворювань (системний червоний вовчак, ювенільний дерматоміозит), а у 11,6% випадків форми клінічних проявів так і залишаються недиференційованими [7].
Іншою формою склеродермії є локалізована, яка представлена ураженням шкіри і може виникати в кількох варіантах [11–15]. За міжнародною класифікацією хвороб ХІ перегляду, ці захворювання визначаються як рубцеві або склерозуючі запальні дерматози, споріднені захворювання погано вивченої етіології, що уражають переважно шкіру та підшкірну клітковину, слизові оболонки та характеризуються різним ступенем склерозу, фіброзу та атрофії шкіри. Найважливішою формою локалізованої склеродермії є морфея (обмежена, бляшечна) — лінійна, генералізована, пансклеротична.
Морфея (шифр за міжнародною класифікацією хвороб ХІ перегляду — EB61) — форма хвороби, коли ураження у вигляді затверділих воскових за кольором бляшок, часто з фіолетовою облямівкою, з’являються на обмеженій частині тіла (зазвичай тулуба). Залежно від локалізації бляшки у пацієнта може відзначатися зменшення додатків шкіри (волосяних фолікулів, потових і сальних залоз). Морфея включає два варіанти — поверхневу і глибоку. Поверхнева є найчастішою формою локалізованої склеродермії у дорослих, у дітей вона діагностується лише в 30% випадків. Глибока форма локалізованої склеродермії діагностується набагато рідше (<5%). Глибока форма може бути без видимих змін поверхневих структур шкіри, характеризується ураженням глибших шарів сполучної тканини, а саме — підшкірної жирової клітковини, фасції, м’язів. Вогнища нерідко розташовані симетрично, переважно на верхніх та нижніх кінцівках. Лінійна форма (EB61.1) — ураження нагадує смугу потовщеної шкіри і зазвичай уражує нижні та/або верхні кінцівки. Ділянки ураження найчастіше розташовані по лініях Блашко (рисунок) [16]. Лінії Блашко, названі в честь німецького дерматолога Альфреда Блашка, є лініями нормального ембріонального розвитку клітин шкіри та слизових оболонок. Ці лінії невидимі в нормальних умовах. Вони стають явними, коли деякі захворювання діагностують відповідно до цих закономірностей, мають V-подібну форму в ділянці хребта, S-подібні завитки на грудях і боках і хвилеподібні форми на голові (див. рисунок). Вважається, що ці лінії відбивають міграцію ембріональних клітин і є різновидом генетичного мозаїцизму. Вони не відповідають анатомічному розташуванню нервової, м’язової, судинної або лімфатичної систем.
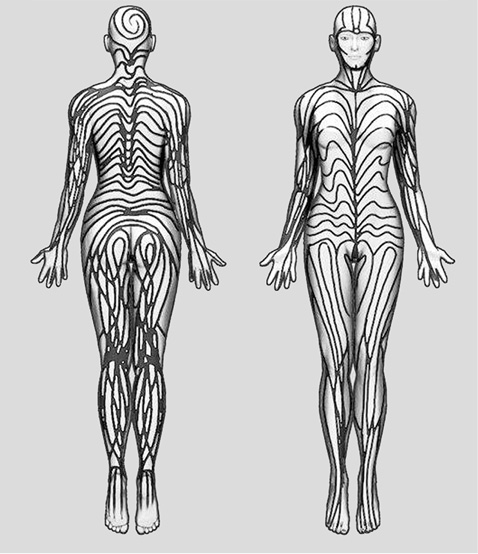
Різновидом лінійної форми є склеродермія за типом en coup de sabre (удар шаблею) (шифр ЕВ61.1), при якому ураження з’являються на скальпі і нагадують за формою шабельні шрами. Станом, спорідненим із типом «en coup de sabre», є прогресуюча атрофія обличчя (синдром Паррі — Ромберга), коли має місце первинна трансформація залучених в патологічний процес підшкірної клітковини м’язів і кісток одного боку голови. Дана форма часто супроводжується супутніми патологічними змінами з боку центральної нервової системи (судоми, головний біль, синкопальні стани), органа зору, зубів, щелеп [17].
Саме лінійна морфея — це форма морфеї, яка зазвичай відмічається в дитинстві або підлітковому віці, в багатьох випадках може бути односторонньою. Для пацієнтів дитячого та підліткового віку важливою особливістю є маніфестація позашкірних проявів лінійної форми у 22–71% випадків. До асоційованих з лінійною формою відносять ураження опорно-рухового апарату — артралгії, артрити, контрактури суглобів (до 20%), позитивність за ревматоїдним фактором (25–40%), міозити, міалгії, м’язові спазми, атрофію м’язів і розвиток вторинного сколіозу; неврологічні порушення (судоми, головний біль, поведінкові порушення), патологію зубощелепної системи дітей зі склеродермією обличчя, особливо при розвитку хвороби у віці до 10 років, гіпоплазію верхньої і нижньої щелепи на ураженому боці при синдромі Паррі — Ромберга [12].
Окремою проблемою є залучення в процес органа зору. Відомо, що ураження очей при локалізованій склеродермії відмічають у 3% хворих, з яких третина не мають склеродермічних уражень на обличчі. Передній увеїт або епісклерит розвивається у 30%, а ураження повік і вій — у 42% педіатричних хворих [13].
Генералізована форма (шифр EB61.Y) — варіант, коли ураження охоплюють декілька частин тіла (принаймні дві анатомічні ділянки) і є набагато більшими та більш дифузними (4 або більше вогнища розміром >3 см в діаметрі). Вогнища часто розташовуються симетрично, але можуть зливатися у більші, і при цьому бляшки можуть мати різні стадії розвитку патологічного процесу — від гіперемії з пігментацією до атрофії.
Пансклеротична склеродермія — вкрай рідкісна, але найтяжча форма локалізованої склеродермії, характеризується розповсюдженим ураженням всіх шарів шкіри тулуба, кінцівок, шкіри обличчя і голови, інтактними залишаються кінчики пальців рук і ніг. Залучення всього тіла без ураження внутрішніх органів допомагає відрізнити пансклеротичну склеродермію від системної. Найчастішим ускладненням є хронічні виразки, які можуть трансформуватися в плоскоклітинну карциному, у багатьох випадках формуються контрактури суглобів з деформацією кінцівок. Зазвичай дана форма відмічається у дітей, є досить торпідною до терапії, швидко прогресує [4–9].
Слід зазначити, що у поняття «склеродермія» в групі запальних дерматозів також включають склерозний лишай (шифр EB60) — хронічний запальний дерматоз невідомої етіології, який характеризується розвитком білих гладких атрофічних бляшок на шкірі вульви та періанальній ділянці у жінок і на передній плоті та головці статевого члена у чоловіків, з можливим ураженням інших ділянок шкіри. Це часто призводить до утворення рубців з порушенням сечовидільної та статевої функції, є фактором підвищеного ризику аногенітальної плоскоклітинної карциноми. Іншими формами є екстрагенітальний склероз з морфеєю (EB60.Y), атрофодермія Пазіні і П’єріні (EE7Y), які також є підставою для диференційної діагностики локалізованої склеродермії.
Епідеміологічні дані щодо ССД неповні, головним чином тому, що вона є рідкісним захворюванням. За даними Європейської групи випробувань і досліджень склеродермії (European Scleroderma Trials and Research — EUSTAR), захворюваність на склероз зростає. Поширеність ССД становить 17,6 на 100 тис. населення, захворюваність становить в середньому 1,4 на 100 тис. населення. Середній вік хворих — 40 років [18–22].
ССД, як і більшість аутоімунних захворювань, набагато частіше відмічається у жінок, ніж у чоловіків, співвідношення захворюваності на ССД у жінок і чоловіків у всьому світі відрізняється 3:1 і 8:1 [22]. Смертність осіб із ССД є значно вищою, ніж у загальній популяції (стандартизований коефіцієнт смертності (SMR) = 2,72). Показано, що через 5 років з моменту встановлення діагнозу виживають 74,9% пацієнтів, при цьому через 10 років — лише 62,5% [23]. Майже 50% випадків смерті пов’язані з серцево-судинними або легеневими розладами, найпоширенішими причинами смерті є ураження легень і розвиток інтерстиціального захворювання легень, пов’язаного із ССД (35% випадків смерті) [23]. У педіатричній практиці ювенільна склеродермія є ще рідшою патологією, захворюваність становить 0,3–3 на 100 тис. з найбільшою частотою у 15–17-річних пацієнтів — 4,3–5,9 на 100 тис. [4]. Дівчатка хворіють в 2,4 раза частіше, але особливістю дитячої склеродермії є однакова частота хвороби у хлопчиків та дівчаток віком до 8 років, серед старших дітей превалюють дівчатка (3:1). Середній вік дебюту становить близько 7 років незалежно від підтипу хвороби.
Етіопатогенетичні аспекти. Загальноприйнято вважати, що головним фактором розвитку склеродермії є генетичні та екологічні предиктори [9]. Сучасні дослідження свідчать, що більшість ідентифікованих генів, які могли б врахувати схильність до розвитку ССД, пов’язані з іншими аутоімунними захворюваннями (так званий спільний аутоімунітет). Вагомим фактором розвитку ССД вважається асоціація локусів HLA DRB1*1104, DQA1*0501 і DQB1*0301 і PTPN22, NLRP1, STAT4 [9]. Також показана роль miR-21 (мікроРНК 21) і miR-29 [24]. Серед факторів навколишнього середовища, які зумовлюють розвиток ССД, найважливішими є інфекційні агенти [25]: цитомегаловірус (ЦМВ) [26], вірус Епштейна — Барр (EBV) [27] і парвовірус B19 [28]. Обговорюється значущість забруднюючих речовин та хімічних агентів, які можуть ініціювати патологічні зміни: кремнеземний пил, органічні розчинники, толуол, ксилол, трихлоретилен і полівінілхлорид [9].
Патогенетична еволюція ССД поєднує судинні, запальні, імунологічні зсуви та порушення згортання крові. Вважається, що першим етапом розвитку захворювання є дисфункція ендотеліальних клітин судин з їх надмірною активацією, синтез чисельних вазомоторних речовин, що впливають на коагуляцію і фібриноліз, проникність судинної стінки та розвиток запальних процесів. Це призводить до порушення співвідношення модуляторів тонусу судинної стінки за рахунок надмірного синтезу ендотеліну та зниженого синтезу оксиду азоту (NO) і простациклінів. Клітинні та молекулярні взаємодії та зміни, які відбуваються в умовах дебюту ССД, є доволі значними, але погано зрозумілими. Доведено участь активованих моноцитів, макрофагів, які поляризуються переважно до клітин М2, дендритних та гладких клітин, CD4+ лімфоцитів (переважно Th2-клітин) та активованих В-лімфоцитів. Підвищується синтез широкого спектру інтерлейкінів (IL-1, IL-4, IL-6, IL-10, IL-13), факторів росту (TGFβ, PDGF, CTGF,VEGF), інтерферону типу I (IFN-α, IFN-β), аутоантитіл або навіть ферментів (аргіназа-1), що викликає надмірну проліферацію клітин внутрішньої оболонки судин і гладком’язових клітин, активацію фібробластів і абсолютно неорганізований синтез позаклітинного матриксу [9]. Перманентність цього стану призводить до накопичення активних форм кисню, гіпоксії та синтезу факторів росту, що призводить до судинного ремоделювання та фіброзу тканин. Відкладення колагену, а також гіалуронової кислоти, глікозаміногліканів і фібронектину утворюють товсту і жорстку сполучну тканину, яка руйнує оригінальну архітектуру і порушує функцію тканин [29].
Особливості маніфестації склеродермії та діагностичні підходи. Класичним клінічним проявом захворювання є наявність уражень шкіри, які відмічаються майже у всіх пацієнтів і проявляються типовою втратою еластичності, сильним відчуттям напруги, які з часом супроводжуються потовщенням та затвердінням. Тяжкість уражень та їх розташування можуть відрізнятися і проявлятися у вигляді обмеженої або дифузної форми ССД [30].
Виділяють три фази розвитку шкірних проявів. Перша, так звана фаза опухлого пальця, пов’язана із загостренням запалення всередині шкіри, в результаті чого виникають неямкові набряки пальців і кистей, свербіж, біль. Супутній тиск на внутрішні структури часто призводить до стискування і проявів нейропатії, випадіння придатків шкіри. Дуже типовим для цієї стадії є виникнення феномену Рейно (періодична ішемія пальців, викликана холодом або емоційним стресом). У другій фазі ураження шкіри в перебігу ССД, так званій пролонгованій, фіброзній фазі, відбувається поступове затвердіння шкіри пальців (склеродактилія), яке може супроводжуватися виразками, рубцями та бактеріальними нашаруваннями інфекції. Характерні численні телеангіектазії (судинні павутинні вени), деформація носа, що нагадує пташиний дзьоб, і набута мікростомія (аномально маленький рот) разом із радіальними борознами навколо рота. Склероз і потовщення разом із вищезгаданими ураженнями спотворюють обличчя пацієнта, часто разом формуючи так званий образ маски. «Фаза пом’якшення шкіри» — це остання та рідкісна стадія розвитку ураження шкіри при ССД. Поверхневі шари шкіри можуть з часом розм’якшуватися, повертаючись у вихідний стан, але залишаються підшкірні фіброзні ураження [30].
Одним із найпоширеніших (близько 90% пацієнтів) клінічних проявів ССД є ураження шлунково-кишкового тракту, які можуть локалізуватися на будь-якій ділянці [9]. У середньому 30–70% симптомів пов’язані з ротовою порожниною, 80–90% супроводжується езофагеальною дисфагією. Зміни моторики і прохідність тонкої кишки (60–80%) і товстої кишки (20–50%) та проблеми з дефекацією, пов’язаною з ураженням прямої кишки, також є досить поширеними симптомами [9]. Ураження шлунково-кишкового тракту розвивається внаслідок судинних уражень, наявності ішемії порушення його моторики. Це призводить до пошкодження іннервації, серед іншого, кишкової стінки та прогресуючого фіброзу м’язової тканини аж до повної атрофії [9, 31].
Респіраторні ураження при ССД передусім включають інтерстиціальне захворювання легень [9, 32] та розвиток легеневої артеріальної гіпертензії, що є наслідком ремоделювання дрібних легеневих судин. Частіше відмічаються у пацієнтів з дифузними формами і є найчастішою причиною смертності при ССД (35% всіх випадків смерті, пов’язаних із інтерстиціальним захворюванням, та 26% — із легеневою гіпертензією). Слід пам’ятати, що рентгенологічне зображення є менш важливим для діагностики легеневих уражень при ССД, оскільки легеневий фіброз виявляється лише на пізніх стадіях ураження. Ультразвукове дослідження може бути корисним для візуалізації ділянок нерівномірного потовщення плеври, але основним методом діагностики легеневих проявів ССД є комп’ютерна томографія високої роздільної здатності [33].
Перший опис кардіальних проявів ССД включав фіброзні ураження коронарних артерій, перикарда та міокарда [9], що в клінічній маніфестації найчастіше представлено міокардитом, розвитком ішемічної хвороби серця, фіброзом міокарда, аномаліями провідної системи, клапанною регургітацією, серцевою недостатністю або захворюванням перикарда та/або ендокарда [34]. Частіші випадки ураження серцево-судинної системи відмічаються при дифузній ССД і асоціюються з наявністю антитіл проти топоізомерази I [35]. Слід зазначити, що залучення в патологічний процес серця відбувається дуже підступно, симптомами цих змін є посилення задишки та набряку. Для оцінки морфології та функції серця дуже корисні методи візуалізації — комп’ютерна томографія, яка може візуалізувати фіброз перикарда та випіт, і магнітно-резонансна томографія (МРТ) для оцінки фіброзу та діастолічної серцевої недостатності [36, 37].
Серцеві та легеневі зміни при ССД дуже часто супроводжуються патологічними змінами сечової системи. За даними дослідників, ураження нирок розвивається набагато швидше, ніж ураження легеневої системи, що пов’язано з підвищенням артеріального тиску у нирковій системі кровообігу порівняно з судинами дихальної системи [9]. Судинний фіброз у нирці дуже швидко призводить до пошкодження ниркових клубочків і порушення клубочкової фільтрації. На жаль, клінічні прояви цих змін стають очевидними лише тоді, коли більша частина ниркової тканини зруйнована. Лише у 50% пацієнтів із ССД відмічають відхилення у лабораторних показниках у вигляді протеїнурії, зниження щвидкості клубочкової фільтрації, підвищення рівня креатиніну. Біопсія нирки свідчить про активну та хронічну тромботичну мікроангіопатію, яка характеризується деструктивною тромбоцитопенією і мікроангіопатичною гемолітичною анемією [38]. Аномалії, виявлені в ниркових артеріях, спричинені проліферацією та ремоделюванням оболонок судин, просвіт артерій суттєво зменшується, що призводить до зниження клубочкової фільтрації. Прогресуючий нирково-судинний фіброз ниркової тканини пояснює значно гірший прогноз у пацієнтів із ССД [39]. Тому необхідними є регулярний контроль функції нирок, повторна еходопплерографія ниркових артерій кожні 6 міс або раз на рік.
Лабораторні тести, які використовуються для діагностики ССД, все ще обмежені і в основному базуються на визначенні аутоантитіл, включених за критеріями ACR/EULAR 2013, тобто антицентромерні антитіла, антитіла до топоізомерази I та антитіла до РНК-полімерази III, які демонструють найвищу специфічність щодо ССД [40]. Є дані стосовно асоціації позитивності за окремими антитілами і клінічними проявами захворювання (табл. 1).
Таблиця 1. Антитіла, виявлені при діагностиці ССД (адаптовано за A. Kowalska-Kepczy´nska, 2022)
| Наявність у сироватці крові пацієнта антитіл | Клінічні прояви |
|---|---|
| Топоізомераза I (анти-Scl 70) | Значне ураження шкіри фіброзними утвореннями;
більша ймовірність серйозних змін органів, особливо легень |
| Ендотеліальні клітини, рецептор PDGF і рецептори ендотеліну | Ендотеліальні клітини, рецептор PDGF і рецептори ендотеліну |
| Центромера (анти-CENP-B, -A, -C або -D) | Помірний ступінь фіброзу шкіри;
нечасте залучення органів; негативна кореляція з виникненням неопластичних уражень |
| U11/U12 RNP (анти-RNPC3) | Вищий ризик тяжких проявів з боку шлунково-кишкового тракту;
вищий ризик неопластичних уражень |
| Фібриларин (анти-U3 RNP) | Вищий ризик легеневої гіпертензії та інших серцевих ускладнень |
| Th/To (анти-Th/To) Th/To (анти-Th/To) | Вищий ризик ураження легень |
| Ендотеліальні клітини, рецептор PDGF і рецептори ендотеліну | Прогресуючі фіброзні зміни |
| Велика субодиниця РНК Pol I (RPA194) | Зниження ризику розвитку раку |
| Цитоплазма нейтрофілів (ANCA) | Вищий ризик легеневих ускладнень;
ниркові ускладнення, підвищення смертності |
При діагностиці ранніх стадій ССД необхідна оцінка тяжкості ураження кожного органу, враховуючи функціональні порушення, визначення N-кінцевого пропептиду натрійуретичного гормону (NT-proBNP) і капіляроскопію нігтьової складки. Специфічні зміни капіляроскопії включають петлі сформованих капілярів, поодинокі гигантські капіляри, чисельні розширені капіляри, поодинокі геморагії, неоангіогенез, ділянки аваскуляризації. Також важливими є визначення легеневої функції та проведення ультразвукового дослідження.
За допомогою ультразвукового дослідження можливо діференціювати активну склеродермічну бляшку від неактивної, верифікувати склеротичну фазу локального прояву склеродермічного процесу, виявити залученість в запальний процес таких структур: вторинний запальний процес в суглобах, залозах (щитоподібна, слинна). Ехосеміотика ураження шкіри включає потовщення дерми, фокальне або дифузне зниження ехогенності дерми, підвищення ехогенності підшкірної клітковини та/або м’яких тканин, гіперваскуляризація дерми та/або підшкірної клітковини. Наявність двох і більше з перших трьох критеріїв є ознакою активної склеродермічної бляшки.
У 2013 р. спільний комітет ACR та EULAR опублікував переглянуті критерії класифікації системного склерозу [1, 2], щоб покращити чутливість широко використовуваних попередніх критеріїв класифікації (табл. 2).
Таблиця 2. Переглянуті критерії класифікації системного склерозу ACR/EULAR
| Пункт | Підпункт(и) | Оцінка* |
|---|---|---|
| Потовщення шкіри пальців обох рук, що тягнеться проксимально до п’ястно-фалангових суглобів (наявності цього критерію достатньо для встановлення діагнозу) | Немає | 9 |
| Потовщення шкіри пальців | Опухлі пальці | 2 |
| Склеродактилія (дистальніше п’ястно-фалангових суглобів, але проксимальніше проксимальних міжфалангових суглобів) | 4 | |
| Ураження кінчиків пальців | Виразки кінчиків пальців |
2 |
| Шрами від кінчиків пальців | 3 | |
| Телеангіектазії | 2 | |
| Аномальні капіляри нігтьової складки | 2 | |
| Легенева артеріальна гіпертензія та/або інтерстиціальна хвороба легень | Легенева артеріальна гіпертензія | 2 |
| Інтерстиціальна хвороба легень | 2 | |
| Феномен Рейно | 3 | |
| Аутоантитіла, пов’язані з системним склерозом (максимальний бал 3): антицентромери (anti-CENP-B, -A, -C, or -D)антитопомераза I (anti-Scl 70)анти-РНК-полімераза III (anti-RNAP III) |
3
3 3 |
*Загальний бал визначається шляхом додавання максимального балу в кожній категорії. Пацієнти із загальним балом, що дорівнює або перевищує 9, класифікуються як такі, що мають певний системний склероз (модифіковано на основі критеріїв класифікації системного склерозу 2013) [6]
Щодо локалізованих варіантів склеродермії, їх дебют зумовлений появою характерних вогнищевих змін на шкірі і переважно є дерматологічною проблемою. У розвитку ділянок склеродермії виділяють три стадії: еритема/набряк, склероз (ущільнення) та атрофія шкіри. У типових випадках захворювання починається з появи на шкірі плям рожевого або жовтувато-білого забарвлення, оточених синьо-пурпурною еритемою (бузкове кільце), або гіперпігментованих плям округлої та/або смугоподібної форми, іноді з явищами набряку. У наступну стадію склерозу на ділянках ураженої шкіри формуються осередки ущільнення кольору слонової кістки з гладкою поверхнею та характерним віскоподібним блиском. Шкіра в місцях ураження погано збирається в складку, на цій ділянці випадає волосся, знижується або відсутнє пото- і саловиділення. Надалі маніфестація індурації шкіри знижується, розвивається атрофія цієї ділянки зі змінами кольору шкіри (гіпер- або гіпопігментації) [4, 5, 6].
Оновлені рекомендації з діагностики та лікування системних хвороб сполучної тканини у дітей містять положення про можливість використання клінічних настанов для дорослих пацієнтів, оскільки клінічні прояви і маркери хвороби ідентичні, незважаючи на вік. Але в щоденній практиці ми маємо усвідомлювати, що в дитячому віці клінічна маніфестація системного захворювання може мати свої особливості, які зумовлені ростовими процесами організму дитини, клітинними взаємодіями в умовах превалювання синтезу і дисбалансу гормонального забезпечення. За результатами широкомасштабного дослідження клінічних та імунологічних проявів у 153 дітей із ССД встановлено особливості її проявів порівняно як з дорослими пацієнтами, так і з лімітованою формою (табл. 3) [14].
Таблиця 3. Частота клінічних проявів ССД в дитячому віці
| Діагностичні критерії ССД | Прояви | Частота у дітей (за даними F. Zulian та співавторів), % | Частота при дифузній ССД, N=1349(за даними EUSTAR), % | Лімітована ССД,N=2101, % |
|---|---|---|---|---|
| Обов’язкові | Проксимальний склероз/індурація | 80 | 100 | 100 |
| Судинні |
Синдром Рейно Дигітальні виразки |
75 | 96
43 |
95
33 |
| Гастроінтестинальні |
Дисфагія, гастроезофагеальний рефлюкс Ураження кишечнику |
Всього
7 |
68
22 |
66
21 |
| Кардіальні | Аритмія, серцева недостатність | 8–15 | 17 | 15 |
| Ниркові | Склеродермічний криз, дебют АГ | 5 (1) | 4 | 1 |
| Легеневі |
Легеневий фіброз Зниження дифузної здатності легень Легенева гіпертензія |
Всього
7 |
53
22 |
34
20 |
| Скелетно-м’язові | Артрит, міозит, крепітація сухожиль | 20 | 37 | 23 |
| Антинуклерні антитіла | 23–73 | 75—90 | ||
| Анти-Scl-70 | 28–34 | 40–45 | 23—61 | |
| Антицентромерні антитіла | 7—8 | 20—30 | 6—47 | |
| Антикардіоліпінові антитіла | 14—15 | 10-25 | ||
Аналізуючи наведені дані, слід зазначити, що в дитячому віці значно рідше відмічаються ураження шлунково-кишкового тракту, легеневі та скелетно-м’язові прояви. Навпаки, залучення нирок у дітей виявляють на тому ж рівні, що й у дорослих, навіть дещо частіше. У той же час ураження внутрішніх органів, включаючи легеневу, серцеву, травну систему, нирки та нервову систему, хоча й розвиваються при ювенільній ССД рідше, ніж у дорослих, пов’язані з гіршим прогнозом. У хворих дітей частіше уражуються органи дихання та шлунково-кишкового тракту, рідко розвиваються захворювання серця, нирок і нервової системи.
У 2012 р. для ювенільної ССД розроблений новий інструмент оцінки тяжкості ювенільного системного склерозу (J4S) (табл. 4) [41]. J4S — це система оцінки, яка включає індекси 9 систем органів, кожна з яких оцінюється за шкалою від 0 до 4 відповідно до тяжкості ураження кожного органу. Показано, що J4S дуже ефективний і надійний у виявленні змін у тяжкості захворювання. Експертна група запропонувала використання J4S у повсякденній практиці, хоча й очікує його валідації у великій когорті пацієнтів.
Таблиця 4. Шкала оцінки тяжкості перебігу ювенільної ССД
| 0Немає | 1Низька | 2Помірна | 3Тяжка | 4Кінцева стадія | Макси-мальнаоцінка | |
|---|---|---|---|---|---|---|
| Загальні | ІМТ ≥ норма
Нв ≥ 115 г/л |
ІМТ до 1-го центилю
Нв 100–114 г/л |
ІМТ до 2-го центилю
Нв 90–99 г/л |
ІМТ до 3-го центилю
Нв 70–89 г/л |
ІМТ до 4-го центилю
Нв 70 г/л |
4 |
| Судинні | Відсутні | Вазодилатація | Дистальні вади | Дигітальні виразки | Дигітальні гангрени | 4 |
| Шкірні | MRSS 0 | MRSS 1–14 | MRSS 15–29 | MRSS 30–39 | MRSS >40 | 4 |
| Скелетні | Немає | Обмеження рухів | Артрит
Деформації сухожіль |
2 | ||
| М’язові | Норма | СMAS 39-51 | СMAS 26-38 | СMAS 13-25 | СMAS 0-12 | 2 |
| ШКТ | Норма | Дистальна езофагеальна гіподинамія | Медіальна або висока езофагеальна гіподинамія | Синдром мальабсорбції | Порушення харчування | 4 |
| Легеневі | DLCO >80%
FVC >80% HRCT норма sPAP >30 mm Hg |
DLCO 70–79%
FVC 70–79% HRCT зміни sPAP 31–45 mm Hg |
DLCO 50—69%
FVC 50–69% HRCT зміни sPAP 46–75 mm Hg |
DLCO 50%
FVC 50% HRCT — фіброз sPAP >75 mm Hg |
Залежність від O2 | 8 |
| Кардіальні | Нормальна ЕКГ
ФВ ЛШ >50% |
Порушення на ЕКГ
ФВ ЛШ 45–49% |
Аритмія
ФВ ЛШ 40–44% |
Аритмія, що потребує лікування
ФВ ЛШ 30–39% |
Стійкі ушкодження
ФВ ЛШ <30% |
8 |
| Ниркові | ШКФ >90 мл/хв | ШКФ 75–89 мл/хв | ШКФ 50–74 мл/хв | ШКФ 10–49 мл/хв | Кінцева стадія ниркової недостатності | 4 |
Примітки. MRSS — modified Rodnan skin score, модифікована оцінка шкіри Роднана; CMAS — Childhood Myositis Assessment Scale — педіатрична шкала оцінки міозиту; DLCO — diffusing capacity for carbon monoxide — дифузійна здатність оксиду вуглецю; FVC — Forced Vital Capacity — життєва ємність легень; HRCT — High-resolution computed tomography — комп’ютерна томографія високої роздільної здатності; sPAP — систолічний тиск в легеневій артерії за даними допплер-ехокардіографії; ФВ ЛШ — фракція викиду лівого шлуночка.
Порівняно з дорослою формою вважається, що ювенільна ССД є менш тяжким захворюванням, рідше уражує внутрішні органи, особливо на момент встановлення діагнозу [41]. Однак дані окремих досліджень підкреслюють наявність підгрупи пацієнтів із швидко прогресуючим перебігом хвороби, яка характеризується ранніми ознаками значного ураження внутрішніх органів. Цей тяжкий перебіг пов’язаний із гіршим результатом і вищим рівнем смертності, що свідчить про необхідність більш агресивного лікування.
Труднощі діагностики ССД у дітей також зумовлені значно меншими серологічними змінами і рідшою презентацією специфічних антитіл. Найбільше значення надається виявленню позитивного загального рівня антинуклеарних антитіл (ANA). Рекомендації щодо менеджменту склеродермічної хвороби у дітей вказують на необхідність проведення усім пацієнтам з ізольованим феноменом Рейно оцінки капілярної складки нігтя та аналіз рівня ANA. Якщо капіляроскопія відхиляється від норми та/або ANA є позитивною, рекомендується подальше спостереження. Оцінка стану дитини за шкалою J4S повинна проводитися кожні 6 міс, пацієнти повинні проходити оцінку легеневої функції, ехокардіографію, визначення функціонального стану нирок і модифіковану оцінку шкірних уражень за Роднаном.
Щодо педіатричної локалізованої склеродермії, як правило, захворювання тривалий час залишається нерозпізнаним: від перших проявів до встановлення діагнозу минає в середньому 11–13 міс, а в 20% випадків — більше 2 років [3, 5, 6]. У 70% пацієнтів при первинному зверненні встановлюється помилковий діагноз, з них в 44% випадків первинним діагнозом є атопічний дерматит [4]. При діагностиці та подальшому спостереженні слід оцінити активність патологічного процесу за наступними клінічними ознаками: поява нових вогнищ (документально підтверджених лікарем) за останні 3 міс; збільшення обсягу вже існуючого ураження (документально підтвердженого лікарем) за останні 3 міс; помірна або виражена еритема; прогресуюче ураження глибоких тканин, виявлене лікарем (за фотографією ділянки ураження в динаміці, при МРТ або ультразвуковому дослідженні); збільшення границі ущільнення ураженої ділянки; прогресуюча втрата волосся на голові, включаючи брови та вії, а також на інших ділянках (документована лікарем); підвищення рівня креатинфосфокінази за відсутності інших змін; виявлення активності захворювання при біопсії шкіри [44].
Основними правилами спостереження дітей із локалізованою формою є наступні. Усіх дітей з підозрою на локалізовану склеродермію слід направляти в спеціалізований дитячий ревматологічний центр. Оцінку активності хвороби обов’язково необхідно проводити із застосуванням шкал та інструментів. Можливо використання інфрачервоної термографії для оцінки активності уражень, але атрофія шкіри іноді дає хибнопозитивні результати. Доцільне ультразвукове дослідження з використанням стандартизованої оцінки та кольорового допплера для оцінки активності захворювання і відповіді на лікування. Усі пацієнти з локалізованою склеродермією під час встановлення діагнозу та подальшого спостереження повинні бути ретельно оцінені з повним обстеженням опорно-рухового апарату, у тому числі скронево-нижньощелепного суглоба. МРТ можна вважати корисним інструментом для оцінки ураження опорно-рухового апарату, його слід проводити у випадках, коли склеродермічне ураження перетинає суглоб. Настійно рекомендується, щоб усім пацієнтам із ураженням обличчя та голови з або без ознак неврологічного ураження було проведено МРТ голови та ортодонтичне та щелепно-лицьове обстеження під час діагностики та лікування. Також під час діагностики у всіх дітей із локалізованою формою склеродермії, особливо з ураженням шкіри обличчя та волосистої частини голови, має бути проведено офтальмологічне обстеження, включаючи скринінг на увеїт.
Лікування склеродермії є непростим завданням, найважливішими принципами якого називають індивідуальність, комплексність, ранній початок адекватної терапії [45, 46]. Існуючі в літературі відомості з лікування ювенільної склеродермії суперечливі, що частково відображає різноманітність форм хвороби з різною поширеністю та глибиною ураження тканин. Довгі роки лікування обмежених форм склеродермії проводили лише шляхом місцевого впливу на шкірний процес, у 60–90-ті роки ХХ ст. стали широко застосовувати Д-пеніциламін, гідроксихлорохін з хорошим ефектом, але ці дані не відповідали критеріям доказової медицини, а з часом накопичувалися відомості про неефективність і погану переносимість зазначеного лікування.
На сьогодні існують чіткі міжнародні узгоджені рекомендації як EULAR [45], так і Європейського товариства дитячих ревматологів (Paediatric Rheumatology European Society — PRES) [46, 47], щодо ведення хворих з ювенільною склеродермією, як системною, так і локалізованою. Основною тенденцією є досить агресивне лікування і ранній його початок за наявності ознак активності хвороби при обох формах захворювання.
Міжнародний комітет із 16 експертів з дитячої ревматології, створений для розробки консенсусних рекомендацій щодо ювенільного системного склерозу, першою позицією в лікуванні ССД залишає системні кортикостероїди (КС). Лікування КС слід розпочинати під час встановлення діагнозу, щоб якомога швидше зменшити вираженість запального компоненту. Хоча в літературі бракує доказів стосовно застосування КС при ССД, існує загальний консенсус щодо показань для активної фази запалення, особливо за наявності міозиту або артриту. Єдиної думки щодо тривалості терапії КС, режиму їх дозування та застосування немає.
Прийом метотрексату (МТХ) або альтернативного хворобомодифікуючого антиревматичного препарату (DMARD) також слід розпочати під час встановлення діагнозу. Згідно з Рекомендаціями EULAR для дорослих пацієнтів МТХ показаний для лікування ураження шкіри, пов’язаного зі склеродермією. Експертний досвід свідчить про раннє застосування МТХ при ССД, особливо за наявності ураження шкіри, суглобів, судин та/або шлунково-кишкового тракту.
Якщо хвороба резистентна до МТХ (погіршення показника J4S через принаймні 6 міс терапії МТХ), слід розглянути можливість додавання або переходу на мофетилу мікофенолат (ММФ). Рекомендації EULAR щодо застосування MMФ у дорослих при склеродермії не визначені, однак окремі дослідження свідчать про позитивний вплив на ураження шкіри та легень із хорошою переносимістю.
Враховуючи відсутність доказів використання імуносупресивної терапії при ювенільній ССД, дитячі ревматологи покладаються на опубліковану літературу щодо дорослих пацієнтів. Рекомендації EULAR для дорослих, взяті з двох рандомізованих контрольованих досліджень (РКД), свідчать про позитивну роль циклофосфаміду при інтерстиціальних захворюваннях легень, пов’язаних із ССД [41]. Незважаючи на його відому токсичність, досвід клінічної практики в європейських педіатричних центрах щодо склеродермії також підтверджує показання до застосування циклофосфаміду у разі ураження легень та/або серця.
Наступною групою препаратів, застосування яких зазначено при ССД, є вазодилататори. Судинорозширювальні засоби часто застосовують у дорослих пацієнтів із склеродермією [45], особливо при вазоспастичних явищах кінцівок. Найпоширенішими вазодилататорами є блокатори кальцієвих каналів (БКК), серед яких основним є ніфедипін. Альтернативою БКК є інгібітори фосфодіестерази 5-го типу (PDE-5i), такі як силденафіл. Експерти припускають, що ілопрост, аналог природного простацикліну (PGI2), може бути показаний для лікування тяжких рефрактерних проявів синдрому Рейно або виразок пальців. Якщо виразки пальців є рефрактерними до БКК і простаноїдів або у випадку легеневої гіпертензії II стадії, слід розглянути можливість застосування бозентану. У дорослих бозентан також рекомендований для профілактики виразок пальців [41].
За наявності швидко прогресуючого або рефрактерного захворювання можна розглядати лікування вибраними категоріями біологічних агентів, незважаючи на недостатні докази в педіатричній популяції. Дані літератури щодо лікування анти-CD20 (ритуксимаб) або анти-IL-6 (тоцилізумаб) дорослих форм ССД підтверджують їх ефективність і відносну безпеку, особливо щодо ураження шкіри та дихальних шляхів, їх застосування в педіатричній практиці пропонується лише на основі останніх публікацій [48].
Нові терапевтичні підходи можуть бути спрямовані на специфічні запальні або профіброзні хемокіни та цитокіни, які, як показано, мають надмірну експресію при склеродермії. Нещодавні звіти свідчать про багатообіцяючі результати трансплантації гемопоетичних стовбурових клітин (ТСКК) при ССД у дорослих і вселяють оптимізм щодо цього терапевтичного підходу у дітей, у яких може бути менше пошкоджень органів після розпізнавання захворювання [41]. Однак відсутність консенсусу стосовно техніки трансплантації та високий ризик пов’язаних з трансплантацією захворюваності та смертності свідчать про необхідність подальших досліджень для оцінки ефективності та безпеки цієї процедури в педіатричній популяції.
Рекомендації щодо лікування ювенільної локалізованої склеродермії включають застосування базисних, імуносупресивних засобів та протизапальних препаратів. Останні опубліковані дані свідчать про ефективність системних КС у поєднанні з MTX у пацієнтів з активною склеродермією, особливо при прогресуючій лінійній формі і генералізованій або пансклеротичній морфеї. Пропонується 2 режими введення: перорально у дозі 1–2 мг/кг/добу (максимально до 60 мг) за преднізолоном протягом 2–4 тиж з подальшим поступовим зниженням дози до повної відміни протягом 4 міс або імпульсне внутрішньовенне введення високих доз метилпреднізолону (30 мг/кг маси тіла, максимально 1 г) з різними схемами введення. Це може бути протягом 3 послідовних днів 1 раз на місяць впродовж 3 місяців, тобто сумарно 9 введень. Або 1 ін’єкція 1 раз на тиждень впродовж 12 тижнів, тобто сумарно 12 введень.
Одночасно зі стартом застосування системних КС при локалізованій склеродермії також слід призначати МТХ або альтернативний DMARD. Експерти рекомендують МТХ як перший етап лікування, він демонструє безпеку та ефективність при пероральному прийомі в режимі 15 мг/м2 на тиждень протягом перших 3 міс в комбінації з ГК «bridge-терапія». В останніх рекомендаціях МТХ призначається із розрахунку 1 мг/кг маси тіла на тиждень, але не більше 25 мг на прийом. Крім того, існує рекомендація щодо призначення МТХ під час встановлення діагнозу, особливо при розповсюджених варіантах. МТХ призначається протягом щонайменше 12 міс при ефективності лікування і переносимості. ММФ в дозі 500–1000 мг/м2 можна застосовувати для лікування тяжкої форми або у випадках, рефрактерних до терапії, або при її непереносимості.
Середньодозова фототерапія UVA1 може бути використана для покращення еластичності шкіри при ізольованих (обмежених) морфейних ураженнях. Використання фототерапії у дітей є тривалим, що призводить до високої кумулятивної дози опромінення та підвищеного ризику потенційних довготермінових ефектів, таких як старіння шкіри та канцерогенез.
Є окремі дані про застосування такролімусу, циклофосфаміду і ряду біологічних препаратів (включаючи блокатори фактора некрозу пухлини або інгібітори IL-6) при резистентних захворюваннях або захворюваннях центральної нервової системи. Також на сьогодні немає доказів високого рівня щодо того, коли припиняти терапію МТХ або іншими імуносупресивними препаратами.
Слід зазначити, що міжнародна спільнота ревматологів наполягає на тому, що ретельний моніторинг стану захворювання та самопочуття пацієнтів має проводитися досвідченою мультидисциплінарною та міждисциплінарною командою, що має досвід в лікуванні склеродермічної хвороби. Ведення таких хворих має відбуватися у спеціалізованих центрах, що є важливим для позитивного клінічного результату. Підкреслюється важливість широкої кооперації спеціалістів і міжнародного обміну даними пацієнтів, створення реєстрів та біобанків, що сприятиме підвищенню стандартів медичної допомоги.
Усі експерти — дитячі ревматологи погоджуються, що програма майбутніх досліджень має включати проспективну перевірку оцінки J4S, використання комп’ютерної томографії високої роздільної здатності (HRCT) в оцінці та моніторингу інтерстиціального захворювання легень та відокремлення можливих біомаркерів, специфічних аутоантитіл та серцево-легеневих маркерів (NT-pro-BNP та ін.). Що стосується лікування, програма майбутніх досліджень повинна включати оцінку безпеки та ефективності MMФ, біологічних агентів і нещодавно відкритих антифіброзних засобів.
Таким чином, імплементація останніх рекомендацій щодо діагностики та лікування локалізованої і системної форми склеродермії у дітей необхідна для підвищення ефективності та правильного лікування, яке може істотно вплинути на віддалений результат, зниження захворюваності та смертності від цієї хвороби. Важливою залишається співпраця дорослих і дитячих ревматологів на засадах доказової медицини для зберігання стратегії лікування і якісного подальшого спостереження хворих дітей.
Список використаної літератури
- 1. van den Hoogen F., Khanna D., Fransen J. et al. (2013) 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. Nov., 65(11): 2737–47.
- 2. van den Hoogen F., Khanna D., Fransen J. et al. (2013) 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis., 72(11): 1747–55.
- 3. Athrea B. (2002) Juvenile scleroderma. Curr. Opin. Rheumatol.; 14(5): 553–61. doi: 10.1097/00002281-200209000-0001315.
- 4. Zulian F. (2017) Scleroderma in children. Best Pract. Res. Clin. Rheumatol.; 31(4): 576–595. doi: 10.1016/j.berh.2018.02.004.
- 5. Herrick A.L., Ennis H., Bhushan M. et al. (2010) Incidence of childhood linear scleroderma and systemic sclerosis in the UK and Ireland. Arthritis Care Res.; 62(2): 213–218. doi: 10.1002/acr.20070.
- 6. Zulian F., Athreya B.H., Laxer R.M. et al. (2006) Juvenile Localized Scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology (Oxford); 45(5): 614–620. doi: 10.1093/rheumatology/kei251.
- 7. Zulian F. (2008) Systemic sclerosis and localized scleroderma in child-hood. Rheum. Dis. Clin. North Am.; 34(1): 239–55.
- 8. Ananyeva L.P., Starovoitova M.N., Shabanova S.Sh. (2014) The main results of cooperation between the V.A. Nasonova Research Institute of Rheumatology and research centers of Europe within the EUSTAR (EULAR Scleroderma Trials and Research group) in systemic sclerosis. Rheumatology Science and Practice; 52(6): 682–688.
- 9. Kowalska-Kepczynska A. (2022) Systemic Scleroderma—Definition, Clinical Picture and Laboratory Diagnostics. J. Clin. Med., 11: 2299. doi.org/10.3390/ jcm11092299.
- 10. Zulian F. (2016) Systemic sclerodermas. In: Petty RE, Laxer RM, Lindsley CB, Wedderburn LR. Textbook of pediatric rheumatology. 7th ed. Elsevier. P. 384–405.
- 11. Foeldvari I. (2015) Update on juvenile systemic sclerosis. Curr. Rheumatol. Rep.;17(3): 18.
- 12. Careta M.F., Romiti R. (2015) Localized scleroderma: clinical spectrum and therapeutic update. An. Bras. Dermatol. Jan-Feb; 90(1): 62–73. doi: 10.1590/abd1806-4841.20152890.
- 13. Zannin M.E., Martini G., Athreya B.H. et al. (2007) Juvenile Scleroderma Working Group of the Pediatric Rheumatology European Society (PRES). Ocular involvement in children with localised scleroderma: a multi-centre study. Br. J. Ophthalmol.; 91(10): 1311–4. doi: 10.1136/bjo.2007.116038.
- 14. Martini G., Foeldvari I., Russo R. et al.(2006) Systemic sclerosis in childhood: clinical and immunologic features of 153 patients in an international database. Arthritis Rheum.; 54(12): 3971–8.
- 15. Vieira Martins M., Azevedo I., Rodrigues C. et al. (2021) Linear scleroderma en coup de sabre-a different clinical presentation. Acta Reumatol. Port., 46: 72–76.
- 16. Holland K.E., Steffes B., Nocton J.J. et al. (2006) Linear scleroderma en coup de sabre with associated neurologic abnormalities. Pediatrics, 117: e132–e136.
- 17. Bielsa Marsol I. (2013) Update on the classification and treatment of localized scleroderma. Actas Dermosifiliogr., 104: 654–666.
- 18. Krasowska D., Rudnicka L., Da ´nczak-Pazdrowska A. et al. (2019) Localized scleroderma (morphea). Diagnostic and therapeutic recommendations of the Polish Dermatological Society. Dermatol. Rev./Przegl. Dermatol., 106: 333–353.
- 19. Hayderi L. (2015) Zosteriform dermatoses-A review. Glob. Dermatol,. 2(4): 163–173. doi: 10.15761/GOD.1000146.
- 20. Wu E.Y., Rabinovich E.C., Torok K.S. et al. (2011) Description of the localized scleroderma subgroup of the CARRAnet. Arthritis Rheumatol.; 63 (Suppl. 10): S787–88.
- 21. Bairkdar M., Rossides M., Westerlind H. et al. (2021) Incidence and prevalence of systemic sclerosis globally: A comprehensive systematic review and meta-analysis. Rheumatology, 60: 3121–3133.
- 22. Hughes M., Pauling J., Armstrong-James L. et al. (2020) Gender-related differences in systemic sclerosis. Autoimmun. Rev., 19: 102494.
- 23. Rubio-Rivas M., Royo C., Simeón-Aznar C.P. et al. (2014) Mortality and survival in systemic sclerosis: Systematic review and meta-analysis. Semin. Arthritis Rheum., 44: 208–219.
- 24. Zhou B., Zuo X.X., Li Y.S. et al. (2017) Integration of microRNA and mRNA expression profiles in the skin of systemic sclerosis patients. Sci. Rep., 7: 42899.
- 25. Radi´c M., Kaliterna D.M., Radi´c J. (2010) Infectious disease as aetiological factor in the pathogenesis of systemic sclerosis. Neth J. Med., 68: 348–353.
- 26. Arcangeletti M.-C., D’Accolti M., Maccari C. et al. (2020) Impact of Human Cytomegalovirus and Human Herpesvirus 6 Infection on the Expression of Factors Associated with Cell Fibrosis and Apoptosis: Clues for Implication in Systemic Sclerosis Development. Int. J. Mol. Sci., 21: 6397.
- 27. Farina A., Peruzzi G., Lacconi V. et al. (2017) Epstein-Barr virus lytic infection promotes activation of Toll-like receptor 8 innate immune response in systemic sclerosis monocytes. Arthritis Res. Ther., 19: 39.
- 28. Zakrzewska K., Arvia R., Torcia M.G. et al. (2019) Effects of Parvovirus B19 In Vitro Infection on Monocytes from Patients with Systemic Sclerosis: Enhanced Inflammatory Pathways by Caspase-1 Activation and Cytokine Production. J. Investig. Dermatol., 139: 2125–2133.e1.
- 29. Allanore Y. (2016) Physiopathologie de la sclérodermie systémique [Pathophysiology of systemic sclerosis]. Med. Sci., 32: 183–191.
- 30. Adigun R., Goyal A., Bansal P. et al. (2021) Systemic Sclerosis. 9 May 2021. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA,.
- 31. Schmeiser T., Saar P., Jin D. et al. (2012) Profile of gastrointestinal involvement in patients with systemic sclerosis. Rheumatol. Int., 32: 2471–2478.
- 32. Wells A.U. (2014) Interstitial lung disease in systemic sclerosis. Presse Med., 43 Pt 2: e329–e343.
- 33. Rutka K., Garkowski A., Karaszewska K. et al. (2021) Imaging in Diagnosis of Systemic Sclerosis. J. Clin. Med., 10: 248.
- 34. Nie L.-Y., Wang X.-D., Zhang T. et al. (2019) Cardiac complications in systemic sclerosis: Early diagnosis and treatment. Chin. Med. J., 132: 2865–2871.
- 35. Meune C., Vignaux O., Kahan A. et al. (2010) Heart involvement in systemic sclerosis: Evolving concept and diagnostic methodologies. Arch. Cardiovasc. Dis., 103: 46–52.
- 36. Dumitru R.B., Bissell L.-A., Erhayiem B. et al. (2021) Cardiovascular outcomes in systemic sclerosis with abnormal cardiovascular MRI and serum cardiac biomarkers. RMD Open, 7: e001689.
- 37. Dumitru R.B., Bissell L.-A., Erhayiem B. et al. (2021) Predictors of subclinical systemic sclerosis primary heart involvement characterised by microvasculopathy and myocardial fibrosis. Rheumatology, 60: 2934–2945.
- 38. Uriarte M.H., Larrarte C., Rey L.B. (2018) Scleroderma Renal Crisis Debute with Thrombotic Microangiopathy: A Successful Case Treated with Eculizumab. Case Rep. Nephrol., 6051083.
- 39. Shanmugam V.K., Steen V.D. (2010) Renal Manifestations in Scleroderma: Evidence for Subclinical Renal Disease as a Marker of Vasculopathy. Int. J. Rheumatol., 538589.
- 40. Utsunomiya A., Oyama N., Hasegawa M. (2020) Potential Biomarkers in Systemic Sclerosis: A Literature Review and Update. J. Clin. Med., 9: 3388.
- 41. La Torre F., Martini G., Russo R. et al. (2012) A preliminary disease severity score for juvenile systemic sclerosis. Arthritis & Rheumatism, 64: 4143–4150.
- 42. Богмат Л.Ф., Никонова В.В. (2019) Ювенильная очаговая склеродермия: клиника, диагностика, современные подходы к терапии (обзор литературы и собственные наблюдения): Zdorov’e Rebenka.; 14(4): 270–277. doi: 10.22141/2224-0551.14.4.2019.174042.
- 43. Охотнікова О.М., Гладуш Ю.І., Мелліна К.В. та ін. (2008) Вісцеральна форма прогресивного системного склерозу (системної склеродермії) у дітей. Клінічна іммунологія. Алергологія. Інфектологія, 5 (16).
- 44. Arkachaisri T., Vilaiyuk S., Li S. et al. (2009) The localized scleroderma skin severity index and physician global assessment of disease activity: a work in progress toward development of localized scleroderma outcome measures. J. Rheumatol. Dec.; 36(12): 2819–29. doi: 10.3899/jrheum.081284.
- 45. Kowal-Bielecka O., Fransen J., Avouac J. et al. (2017) Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis.; 76:1327–1339.
- 46. Foeldvari I., Culpo R., Sperotto F. et al. (2021) Consensus-based recommendations for the management of juvenile systemic sclerosis. Rheumatology (Oxford). 6; 60(4): 1651–1658. doi: 10.1093/rheumatology/keaa584.
- 47. Zulian F., Culpo R., Sperotto F. et al. (2019) Consensus-based recommendations for the management of juvenile localised scleroderma. Annals of the Rheumatic Diseases; 78: 1019–1024.
- 48. Zulian F., Dal Pozzolo R., Meneghel A. et al. (2020) Rituximab for rapidly progressive juvenile systemic sclerosis. Rheumatology; 59: 3793–7.
Leave a comment