СИНДРОМ ШНІЦЛЕР: ЗНАЙОМА КРОПИВ’ЯНКА ПРИ НЕЗНАЙОМИХ АУТОЗАПАЛЬНИХ ЗАХВОРЮВАННЯХ. ДОСВІД ЛІКУВАННЯ ДИТИНИ ІЗ СИНДРОМОМ ШНІЦЛЕР У ПЕДІАТРИЧНОМУ ВІДДІЛЕННІ КНП «ОКДЛ КОР» М. КРОПИВНИЦЬКИЙ
Мірошниченко М.В., Шишканова Н.В., Іванова В.Г.
Резюме. Синдром Шніцлер (СШ) — це аутозапальне захворювання, асоційоване з моноклональною гамапатією IgM (рідше IgG), що проявляється хронічним уртикарним висипом, ознаками системного запалення у вигляді лихоманки, артралгією та/або болем у кістках, а також підвищенням запальних показників крові. Незважаючи на те що захворювання відоме з 1972 р., коли воно було вперше описано Ліліан Шніцлер, ця патологія рідко діагностується у світі [1]. Найвища ефективність лікування виявлена при застосуванні інгібіторів інтерлейкіну-1 (IL-1) [2, 3], проте, на жаль, слід зазначити, що на фоні припинення прийому ліків відмічалося повернення симптомів [4], тому хворі потребують довічного лікування. Мета: підвищити обізнаність медичної спільноти щодо клінічних проявів, методів діагностики та лікування СШ. Ознайомити з клінічними результатами застосування інгібіторів IL-1 у лікуванні даного синдрому у дитини в педіатричному відділенні КНП «Обласна клінічна дитяча лікарня (ОКДЛ) Кіровоградської обласної ради (КОР)» м. Кропивницький. Об’єкт і методи: оброблено сучасні літературні дані. Представлено клінічний випадок СШ у дитини, діагностований на підставі Страсбурзьких критеріїв, на базі ОКДЛ КОР. Проаналізовано ефективність лікування СШ інгібітором IL-1 та побічні реакції протягом року терапії. Результати: на фоні лікування інгібітором IL-1 протягом 1 року у пацієнтки відмічалося значне покращення загального самопочуття: зменшення вираженості шкірних проявів аж до повного їх зникнення, відсутність лихоманки, артралгій/осалгій, які були частиною симптомокомплексу на момент встановлення діагнозу. На фоні введення препарату відзначалися такі побічні реакції: місцеві у вигляді почервоніння та ущільнення місця ін’єкції, дворазово — системні прояви у вигляді ларингоспазму в поєднанні з вираженою кропив’янкою, які купірувалися антигістамінними препаратами. Висновки: СШ є рідкісним захворюванням, що виявляється недостатньо. Висока настороженість щодо СШ необхідна при обстеженні пацієнтів із хронічними дерматозами, подібними до кропив’янки, періодичною лихоманкою та артралгіями. Для уточнення діагнозу слід провести додаткові аналізи — електрофорез сироваткових білків з імунофіксацією. Захворювання важливо вчасно розпізнати, оскільки СШ асоційований із появою злоякісних новоутворень. У близько 20% пацієнтів в середньому через 7,6 року після появи симптомів розвивається лімфопроліферативний розлад. Таким чином, вони потребують постійного та тривалого спостереження. Інгібітори IL-1 надзвичайно ефективні в зменшенні вираженості симптомів і вважаються терапією першої лінії.
DOI: 10.32471/rheumatology.2707-6970.91.17528
УДК 616 (614.4 та 616.5)
Синдром Шніцлер (СШ) — це рідкісне набуте аутозапальне захворювання, що проявляється хронічним уртикарним висипом, ознаками системного запалення у вигляді лихоманки та лімфаденопатії, артралгіями та/або болем у кістках, а також підвищенням запальних показників крові, асоційованим з моноклональною гамапатією IgM (рідше IgG).
Епідеміологія: СШ є рідкісним захворюванням, яке виявляється недостатньо. На підставі наявних даних відомо, що в середньому чоловіки хворіють у 1,5 раза частіше, ніж жінки [1, 5], більшість випадків припадає на осіб європеоїдної раси [6]. Медіаною віку початку захворювання є в середньому 51 рік (на підставі дослідження 174 пацієнтів), а відтермінування у встановленні правильного діагнозу зазвичай перевищує 5 років [6]. Вік наймолодшого пацієнта з СШ, який описаний у проаналізованих ресурсах, — 13 років [7].
Етіологія та патогенез. Провідну роль у патогенезі СШ відіграють запальні та прозапальні цитокіни, головним чином інтерлейкін-1b (IL-1b), який виробляється під впливом інфламасоми NLRP3 (NLR family pyrin domain containing 3), також відомого як кріопірин [8]. Також у пацієнтів із СШ у крові виявлено підвищення рівня IL-1a, IL-1b, IL-6, IL-18 та фактора некрозу пухлини (TNF) порівняно з контрольною групою [6, 9, 10]. Проте саме IL-1b відповідає за запальні прояви СШ, такі як лихоманка та підвищення запальних маркерів — швидкість осідання еритроцитів (ШОЕ) та С-реактивного білка (СРБ).
Подібність клінічного фенотипу СШ та кріопіринасоційованого періодичного синдрому (CAPS) і хороша терапевтична відповідь на інгібітори IL-1 свідчить про активацію інфламасоми NLRP3, яка може бути відповідальною за надмірне виробництво IL-1. При CAPS ця гіперпродукція виникає за рахунок мутаційних змін у однойменному гені (NLRP3). На підставі цього висувалися припущення, що і при СШ будуть виявлені генетичні зміни. Однак такі мутації в гені NLRP3 не відмічені у пацієнтів із класичним СШ [11, 12].
Однак гіперпродукція IL-1 і переважний терапевтичний ефект застосування інгібіторів IL-1 вказують на те, що аутозапалення при СШ опосередковується головним чином саме завдяки IL-1. Погано вивчено, що є основним клітинним джерелом IL-1, але доцільно вважати, що нейтрофіли та гладкі клітини в пошкодженій шкірі можуть зумовлювати надмірне вироблення IL-1 [13].
Однак точна причина гіперпродукції IL-1 при класичному СШ все ще невідома.
Парапротеїнемія у вигляді моноклональної гамапатії — це друга важлива складова у розвитку СШ. Моноклональні гамапатії (МГ) — це група захворювань, що характеризуються зростанням одного клону плазмоцитів, який виробляє гомогенний, тобто моноклональний білок (М-протеїн або парапротеїн), що складається з цілої молекули імуноглобуліну (2 важких ланцюгів того ж класу і 2 легких ланцюгів того ж типу) або з самого легкого ланцюга цього ж типу, рідше — тільки з тяжких ланцюгів такого самого типу (від цього залежить підтип гамапатії — лямбда, каппа і т.ін.)
Незважаючи на те що МГ є обов’язковим критерієм для встановлення діагнозу СШ, досі майже нічого невідомо про роль парапротеїнемій у патогенезі цього синдрому. Цікавим є те, що лікування інгібіторами IL-1, яке призводить до значного зменшення вираженості запалення та клінічної симптоматики СШ, жодним чином не впливає на гамапатію [12, 14], отже, можна припустити, що IL-1 не відповідає за клонування глобулінів. Проте на це можуть впливати інші фактори.
Таким фактором може бути IL-18, інший член сімейства IL-1. У дослідженні P. Migliorini та співавторів (2019) у пацієнтів із СШ виявлено підвищення рівня IL-18 порівняно з контрольною групою [10]. Отже, можна припустити, що певний стимул/стимули одночасно активують інфламасому NLRP3 і В-клітину (рис. 1).
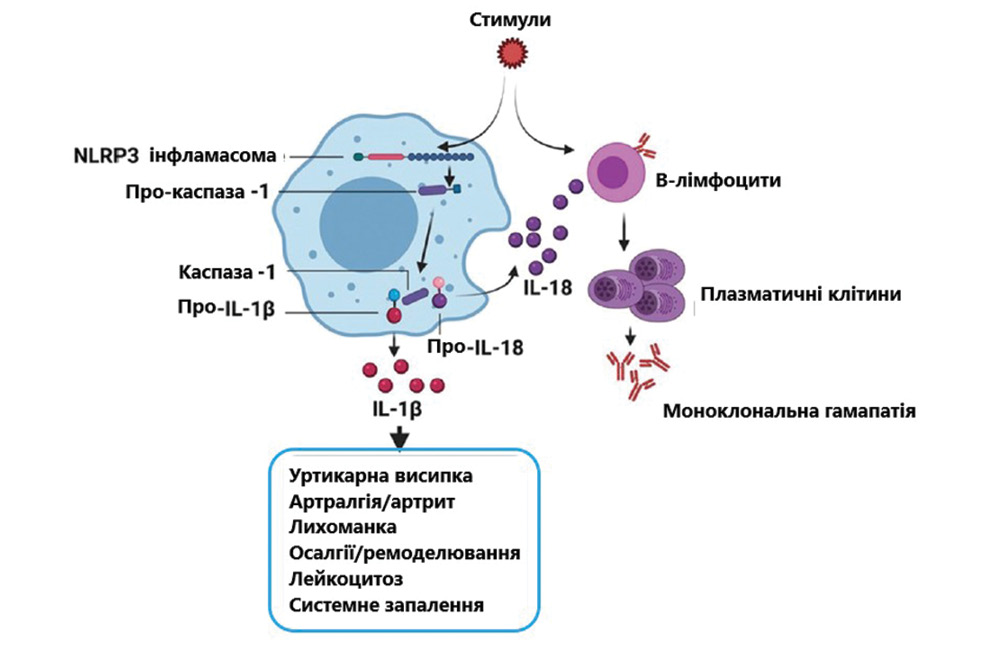
За допомогою NLRP3 активується каспаза-1 (вона ж інтерлейкін-1-перетворювальний фермент), вивільняються IL-1b та IL-18. Тоді як IL-1b викликає системне запалення, IL-18 діє на активовані В-клітини та ініціює у них клонування глобулінів, що і призводить до МГ невизначеного значення (Monoclonal gammopathy of undetermined significance — MGUS). Продемонстрована роль IL-18 у зростанні В-клітин і виробленні антитіл — патогенспецифічного IgM [15]. Також важливо відмітити, що IL-18 бере безпосередню участь у розвитку проліферативних розладів В-клітин [16].
Клінічні прояви СШ:
1. Шкірний висип у більшості пацієнтів зазвичай відмічається як перший клінічний симптом СШ та може на декілька років передувати появі лихоманки [6]. Візуально він має блідо-рожевий або червоний колір, складається з макул (плоскі ураження) або злегка підвищених над поверхнею шкіри папул і бляшок. Його часто характеризують як уртикарний, проте слід зазначити, що гістологічно він належить до нейтрофільних дерматозів. При біопсії типового елементу на відносно ранній стадії у дермі відмічається нейтрофільний інфільтрат [17, 18]. Нейтрофільний уртикарний дерматоз (НУД) — це обов’язковий критерій для СШ, він має відмічатися у всіх пацієнтів. За умови наявності інших ознак СШ, але без шкірного висипу, діагноз пацієнта може бути охарактеризований як Шніцлерподібний синдром [19].
Зазвичай висип відмічається на шкірі тулуба та кінцівок, рідше — в ділянці голови. Окремі ураження зберігаються менше 24–48 год. Лише в 21% випадків ураження з часом викликає свербіж, часто ураження шкіри пов’язані з відчуттям печіння, а не свербежу. Ангіоневротичний набряк виявляють рідко. [4] Частота загострень варіює: у пацієнтів можуть виникати щоденні спалахи протягом місяців або років або ремісія від днів до тижнів. Однак у хворих, що не отримують лікування, періоди без висипу вкрай рідко перевищують місяць.
При проведенні імунофлуоресцентних досліджень у близько 30% пацієнтів можна виявити відкладення аутоантитіл, головним чином IgM, навколо поверхневих судин шкіри [20, 21, 22]. Роль цих антитіл залишається нез’ясованою, хоча висловлювали припущення, що вони можуть викликати місцеву запальну реакцію, яка зумовлює ураження шкіри [23].
2. Лихоманка є другим за поширеністю симптомом та відмічається більше ніж у 70% пацієнтів. Вона не має чіткої періодичності, а частота нападів варіює від щоденних до декількох разів на рік, може сягати вище 40 °С. Відчуття ознобу буває рідко, проте загальна слабкість часто відмічається під час спалахів лихоманки. У літературі описані випадки розвитку лихоманки через проміжок часу 1–14 років від виникнення шкірних проявів, хоча частіше вони відмічаються майже одночасно. У деяких пацієнтів лихоманка реагує на нестероїдні протизапальні препарати (НПЗП) та/або стероїди, і зазвичай повністю контролюється IL-1 інгібіторами [4, 24].
3. Ураження опорно-рухового апарату є ще однією основною ознакою захворювання і відмічається майже у 80% пацієнтів [4, 6]. Найчастіше виявляють осалгії, проте можуть виникати і артралгії, іноді — повністю розвинений артрит. Руйнування та/або деформації суглобів не відмічалося. Біль переважно турбує в клубових та гомілкових кістках, рідше — у стегнових кістках, хребті, передпліччі та ключиці [25]. Під час візуалізації органічне ураження кісток виявлено у 30–40% пацієнтів [26]. Остеоконденсація є найчастішою рентгенологічною знахідкою, а найпоширенішою рентгенологічною картиною є склеротичне ураження кісткового мозку з кортикальним гіперостозом дистального відділу стегна та проксимального відділу великогомілкової кістки, а також відсутністю ознак злоякісності [6].
Лише 38% кісткових аномалій виявлені за допомогою звичайної рентгенограми, а решта потребували використання інших методів (ядерної сцинтіографії, позитронно-емісійного сканування, сканування галію, магнітно-резонансної томографії або комп’ютерної томографії). З огляду на високу чутливість ядерна сцинтиграфія може використовуватися як початковий метод скринінгу [27].
4. Лімфопроліферативний синдром: збільшення лімфатичних вузлів відмічається у близько 45% пацієнтів, а збільшення печінки або селезінки — приблизно у третини пацієнтів [7, 20]. Пальпуються збільшені лімфовузли в пахвових і пахових ділянках, іноді в шийному відділі. Ці лімфатичні вузли можуть бути множинними, постійними та розміром до 2–3 см, тому часто диференційний діагноз проводиться з лімфомою, проте біопсія у випадку СШ показує неспецифічне запалення.
5. Нейропатія також іноді відмічається у пацієнтів із СШ, в середньому її частота близько 7%, проявляється зазвичай у вигляді симетричної сенсорної полінейропатії [6, 28].
6. Зменшення маси тіла, що відмічається у близько 15% випадків.
Зміни лабораторних показників:
- майже завжди відмічається підвищення сироваткових маркерів системного запалення, таких як ШОЕ і СРБ;
- лейкоцитоз, зазвичай нейтрофільоз, виявляється у ¾ хворих, а анемія — у ⅔ [6];
- парапротеїн (або МГ) є ще одним обов’язковим критерієм для встановлення діагнозу СШ. Підтип IgM-каппа виявляють у 85% хворих, кількість випадків IgG-гамапатії — значно менша (близько 7%) [6]. Слід зазначити, що в усіх дослідженнях до 2013 р. IgG не був включений до критеріїв діагностики СШ і взагалі не брався до уваги при статистичних підрахунках. Тож частка пацієнтів із гамапатією IgG може бути вищою [1, 6]. Цікаво, що у деяких пацієнтів відмічалася біклональна гамапатія (у 7 випадках з 281 досліджених, що проведені Heleen D. de Koning [6]). У 2 із цих випадків виявлена МГ IgA на додаток до гамапатії IgM κаппа. У 1 пацієнта відмічено 3 різних М-білка IgM. У 89% випадків каппа був підтипом легкого ланцюга. Білки Бенс-Джонса у крові або у сечі виявлені в 23% випадків, у яких вони були оцінені;
- підвищення кістково-лужної фосфатази як маркера ураження кісткової тканини;
- анемія, яка іноді може бути дуже тяжкою та симптоматичною [20, 21], розвивається за рахунок хронічного запального процесу у близько 60% пацієнтів [6].
На даний момент у світі найбільш масштабним статистичним та аналітичним дослідженням даного синдрому є оглядова стаття Heleen D. de Koning, у якій проаналізовано 281 випадок СШ [6], підсумовані клінічні особливості симптоматики, ефективність терапії.
Усі пацієнти для вищеописаного дослідження відповідали Страсбурзьким критеріям діагностики (табл. 1) СШ [29], які розроблені на основі критеріїв, запропонованих D. Lipsker та співавторами [20] у 2001 р. Єдиною відмінністю є те, що Страсбурзькі критерії є більш розширеними, оскільки у них був доданий IgG-асоційований варіант СШ (2013 р.).
Таблиця 1. Страсбурзькі критерії діагностики
| Обов’язкові критерії | Додаткові критерії |
|
Хронічний уртикарний висип+ Моноклональна гамапатія IgM або IgG |
Повторна лихоманка* Об’єктивні ознаки аномального ремоделювання кісток з/без осалгій† Нейтрофільні інфільтрати при шкірній біопсії‡ Лейкоцитоз та/або підвищення СРБ§ |
| Точний діагноз | Вірогідний діагноз |
| Є 2 обов’язкових критерії та як мінімум 2 додаткових критерії (якщо IgM), або 3 додаткових критерії (якщо IgG) | Є 2 обов’язкових критерії та як мінімум 1 додатковий критерій (якщо IgM), або 2 додаткових критерії (якщо IgG) |
*Критерій вважається дійсним за умови об’єктивного вимірювання. Має бути >38 °C без інших можливих причин. Виникає зазвичай — проте не обов’язково — разом із шкірним висипом. †Оцінюється на підставі кісткової сцинтіографії, МРТ або підвищення кістково-лужної фосфатази. ‡Зазвичай описується як «нейтрофільний уртикарний дерматоз», відмічається відсутність фібриноїдного некрозу та значного набряку шкіри. §Нейтрофіли >10 000/mm3 та/або СРБ >30 mg/l.
Важливо відмітити, що у дослідженні Cong-Qiu Chu описані 3 випадки, у яких пацієнтам з усіма ознаками класичного СШ, за винятком парапротеїнемії, встановлений діагноз «хронічний уртикарний висип без моноклональної гамапатії» [9]. Проте через певний час у них розвинулася МГ IgM, що відповідало усім необхідним критеріям для встановлення діагнозу СШ [30, 31, 32]. Ці випадки підкреслюють, що уртикарний шкірний висип та інші клінічні фенотипи можуть передувати розвитку МГ. Обов’язковими є тривале спостереження та періодичне обстеження на гамапатію пацієнтів з високою підозрою на СШ.
Диференційний діагноз
Диференційна діагностика СШ проводиться з широким спектром захворювань і ґрунтується на поєднанні клінічних, біологічних і рентгенологічних даних, а також на виключенні іншої причини. Нижче наведено основні нозології, з якими СШ має бути диференційований (табл. 2).
Таблиця 2
| Нозологія, з якою проводиться диференційний діагноз | Схожі ознаки | Відмінні ознаки |
|---|---|---|
| CAPS (кріопіринасоційований періодичний синдром) | Рекурентна лихоманка, артралгія, уртикарний характер висипу, підвищення маркерів запалення та лейкоцитоз, хороша відповідь на терапію інгібіторами IL-1 | Холодозалежні прояви, наявна мутація гена NLRP3, парапротеїнемія відсутня, зазвичай розвивається у дитячому віці |
| Ювенільний артрит з системним початком/хвороба Стілла у дорослих | Рекурентна лихоманка, артралгія, підвищення маркерів запалення та лейкоцитоз, лімфаденопатія, гепатоспленомегалія | Висип зазвичай макулопапулярний, високі рівні феритину у сироватці крові, парапротеїнемія відсутня |
| Системний червоний вовчак | Рекурентна лихоманка, артралгія, уртикарний характер висипу, підвищення маркерів запалення та лейкоцитоз, лімфаденопатія | Характерний висип у вигляді «метелика», парапротеїнемія відсутня, артралгії не відмічаються |
| Макроглобулінемія Вальденстрема | Наявна парапротеїнемія | Рівні моноклональних глобулінів значно вищі (зазвичай >10 000 мг/л), лімфопроліферативні зміни при біопсії кісткового мозку |
| Хронічна спонтанна/індукована кропив’янка (CSU/CIU) (без моноклональної гамапатії) | Уртикарний характер висипу | Більш виражений свербіж, невиражені запальні зміни, висип несиметричний, триває менший проміжок часу, гістологічно — більш виражений дермальний набряк, парапротеїнемія відсутня |
Слід зазначити, що у дослідженні Gilson та співавторів запропоновано вважати виражений та швидкий терапевтичний ефект після введення анакінри ще одним критерієм на користь СШ при диференційній діагностиці [34].
Лікування
Препаратом вибору для лікування СШ є інгібітор рецепторів IL-1 (IL-1Ra) анакінра. Її висока ефективність відмічалася у 94% пацієнтів [6], а досить виражена відповідь після першого прийому препарату зафіксована вже в перші 48 год [9]. Високу ефективність продемонстрував канакінумаб (aнти-IL-1β антитіла) (ефективний у 91% пацієнтів). Менш дієвим виявився тоцилізумаб (aнти-IL-6 антитіла): хороша терапевтична відповідь відмічалася лише у 75% хворих [6].
Рекомендована стартова доза IL-1Ra становить 100 мг/добу [3, 6, 7, 12, 24, 29, 33, 34, 39] з подальшим можливим її зниженням. Потім можна спробувати знижувати дозу до мінімально можливої, яка контролює симптоми. Цього можна досягти, вводячи 100 мг через день або, якщо ознаки повторюються, наступного дня, вводячи кожні 36 год або лише частину шприца щодня (знижуючи введену дозу) [29]. Слід зазначити, що анакінра лише зменшує вираженість запалення, але не лікує хворобу. Симптоми незмінно повертаються у термін від 1 до (рідко) 6 днів після відміни препарату. Основним побічним ефектом є еритематозна реакція в місці ін’єкції, також відмічаються більш часті інфекції верхніх дихальних шляхів. Оскільки на фоні прийому анакінри може виникати нейтропенія, рекомендується щоквартальний розгорнутий аналіз лейкоцитів з коротшими інтервалами протягом перших тижнів після початку лікування. У літературі описано 2 випадки, коли після виникнення нейтропенії на фоні лікування терапія IL-1Ra була призупинена, а в подальшому — успішно реіндукована [35]. Оскільки анакінра потребує болючих щоденних ін’єкцій, необхідні препарати більш тривалої дії. Канакінумаб виявився ефективною та безпечною альтернативою, але не був запропонований для лікування СШ [36].
Нижче наведено схему лікування СШ залежно від вираженості клінічних симптомів, запропоновану A. Simon та співавторами (2013) (рис. 2).
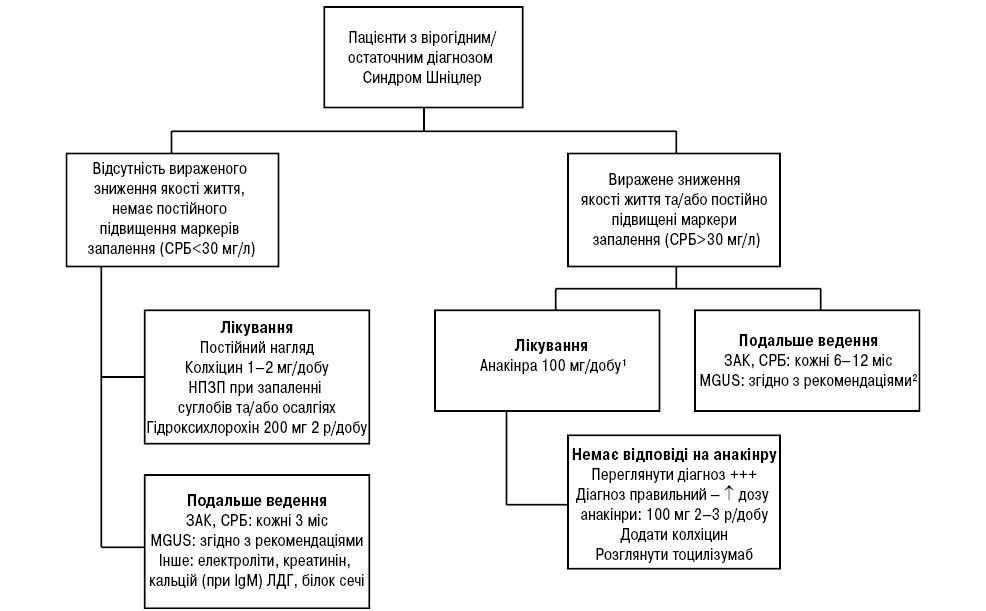
Перебіг хвороби та ускладнення
СШ — це хронічне захворювання, вираженість симптомів якого залежить від належної терапії. При щоденному отриманні інгібіторів IL-1 можна досягнути та підтримувати довготривалу медикаментозну ремісію. Зазвичай після припинення терапії анакінрою симптоми повертаються протягом 1–6 діб [6].
Описаний єдиний випадок класичного СШ, який відмічали протягом більше 8 років без отримання лікарських засобів, а в подальшому перейшов у спонтанну ремісію [4, 37].
Ризик виникнення онкогематологічних захворювань у пацієнтів із СШ є досить високим за рахунок парапротеїнемії. Базуючись на дослідженнях de Koning [6], лімфопроліферативні захворювання розвинулися у 35 пацієнтів із 281 досліджених випадків СШ (12,5%), у середньому через 8 років спостереження з моменту початку хвороби. У ⅔ з вищеописаних пацієнтів виявлена макроглобулінемія Вальденстрема, а у більшості інших випадків — лімфома. Проте кількість пацієнтів із СШ, у яких розвинулося лімфопроліферативне захворювання, може бути більшою, оскільки багато випадків описано протягом короткого періоду після встановлення діагнозу СШ, тож важко робити висновок щодо можливих відтермінованих наслідків. Проте цікаво, що з усіх пацієнтів, які отримували анакінру, згідно із дослідженням de Koning лише у одного пацієнта розвинулася макроглобулінемія Вальденстрема [6].
Амілоїдоз як ускладнення виникає досить рідко. У вищезазначеному дослідженні з 281 пацієнтів лише у 6 випадках (2%) розвинувся А-амілоїдоз [6]. Слід нагадати, що амілоїд-А в сироватці крові (SAA) — це білок гострої фази запалення. Терапія інгібіторами IL-1 контролює запалення при СШ, а отже, ймовірно зумовлює зниження ризику виникнення амілоїдозу.
Нижче наведений клінічний випадок пацієнта з СШ, який лікувався у педіатричному відділенні № 2 КНП «ОКДЛ КОР» м. Кропивницький.
Дівчина Д., 2004 р.н. З анамнезу відомо, що захворювання почалося у 2019 р., коли у пацієнтки вперше з’явився уртикарний висип на тілі, що більше локалізувався на кінцівках, менше — на тулубі та животі. Восени 2019 р. у зв’язку з вищеописаними скаргами звернулася до алерголога, знаходилася на стаціонарному лікуванні у відділенні алергології КНП «ОКДЛ КОР» з діагнозом «алергічний дерматит. Хронічний гастродуоденіт». В аналізах крові відмічався лейкоцитоз (14 г/л), підвищення ШОЕ (59 мм/год) та СРБ (48 мг/л). У подальшому протягом 2020 р. скарги на наявність уртикарного висипу на тілі та кінцівках набули постійного характеру, проте у зв’язку з умовно задовільним загальним станом дитина за медичною допомогою не зверталася, лікувалася симптоматично (антигістамінні препарати). У січні 2021 р. стан дитини погіршився: підвищилася температура тіла до 38,5 °С, з’явилися біль у м’язах, суглобах, набряк міжфалангових суглобів пальців рук, їх почервоніння, ломота в тілі на фоні лихоманки, виражена слабкість, зберігався уртикарний висип на кінцівках та тілі, що змінював своє місцерозташування, везикулярний висип на лівій половині носа та над верхньою губою, відмічався періодичний кашель. У зв’язку зі скаргами хвора госпіталізована в інфекційне відділення КНП «ОКДЛ КОР», де була на стаціонарному лікуванні з 11.02.2021 до 01.03.2021 р. з діагнозом «герпетична інфекція. Атопічний дерматит. Хронічна кропив’янка». В аналізах крові відмічалося підвищення ШОЕ 55 мм/год, СРБ 24мг/л. Була консультована кардіоревматологом, для виключення ревматологічної патології призначено дообстеження: антинуклеарні антитіла (ANA) методом IFT — негативно, антитіла до двоспіральної ДНК — негативно. Виписана з поліпшенням стану для подальшого амбулаторного нагляду та лікування, проте на момент виписки за словами дитини уртикарний висип зберігався.
У зв’язку зі збереженням скарг на періодичну лихоманку до фебрильних цифр, неприємні відчуття у м’язах, суглобах, набряк міжфалангових суглобів пальців рук, їх почервоніння, ломоту в тілі на фоні лихоманки, виражену слабкість, уртикарний висип на кінцівках та тілі, що змінює своє місцерозташування, 05.05.2021 р. повторно госпіталізована у педіатричне відділення КНП «ОКДЛ КОР», м. Кропивницький.
Об’єктивно: на момент госпіталізації загальний стан середнього ступеня тяжкості за рахунок лихоманки, загальнозапальних змін, артралгії. На шкірі — висип уртикарного характеру, більше — на тулубі, руках, гомілках та стегнах, стопах, блідо-рожевого кольору (рис. 3), періодично зникає, висип збільшується на вечір, має тенденцію до міграції, також відмічаються набряклість та почервоніння міжфалангових суглобів пальців рук. Пальпуються множинні збільшені передньошийні та задньошийні лімфовузли справа, розміром до 2 см, рухомі, не з’єднані з навколишніми тканинами, безболісні при пальпації.
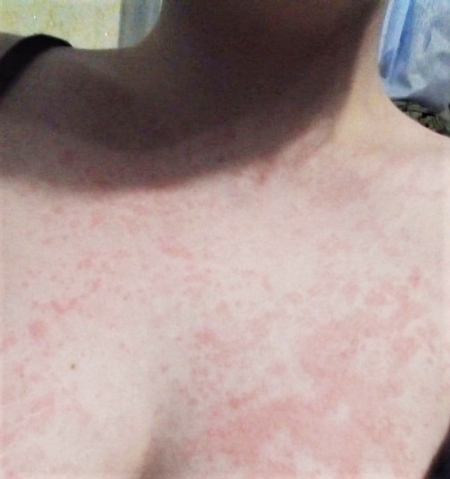
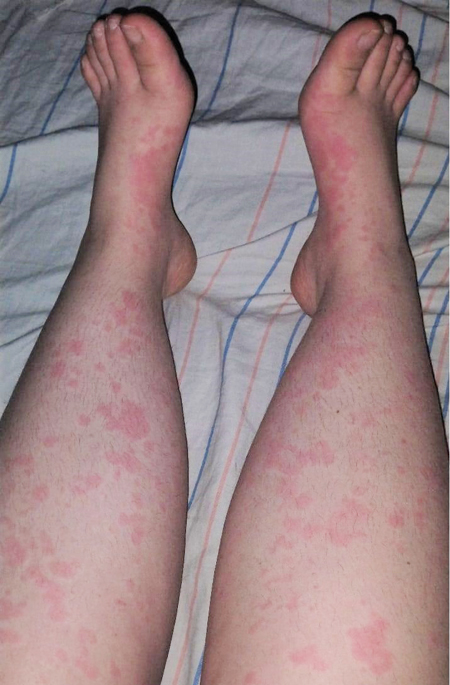
В аналізах крові зберігалися виражені запальні зміни:
| Дата | ||||
|---|---|---|---|---|
| 05.05.2021 | 14.05.2021 | 09.06.2021 | 22.06.2021 | |
| Hb | 104 Г/л | 96 Г/л | 109 Г/л | 90 Г/л |
| Лейк. | 14,5 Г/л | 11,3 Г/л | 8,7 Г/л | 8,2 Г/л |
| ШОЕ | 64 мм/год | 57 мм/год | 42 мм/год | 46 мм/год |
| Еоз. | 0% | 6% | 4% | 0% |
| Пал. | 1% | 11% | 3% | 2% |
| Сегм. | 77% | 65% | 71% | 87% |
| ЛДГ | 606 Од/л | 770 Од/л | – | – |
| КФК | 42 Од/л | 35 Од/л | – | – |
| СРБ | 96 мг/л | 48 мг/л | 12 мг/л | 48 мг/л |
Періодично відмічалися токсична зернистість еритроцитів, анізо- та пойкілоцитоз.
Проводився такий диференційний діагноз: системний червоний вовчак, ювенільний ідіопатичний артрит (системна форма), онкогематологічні захворювання, ВІЛ-інфекція, СШ, CAPS.
Додаткові методи дослідження:
- ультразвукове дослідження (УЗД) лімфатичних вузлів шиї: множинні збільшені передньо-шийні та задньошийні л/в справа;
- УЗД нирок: без органічних змін;
- мультиспіральна комп’ютерна томографія (МСКТ) органів грудної клітки, органів черевної порожнини, малого таза: КТ-ознаки генералізованої лімфаденопатії. Помірна спленомегалія. Гіподенсивне вогнище у S3 печінки (кіста?). Кістоподібне утворення лівого яєчника;
- стернальна пункція: патологічних змін не виявлено;
- полімеразна ланцюгова реакція (ПЛР) (віруси герпесу 1-го, 2-го, 6-го типів) — негативно;
- ехокардіографія (ехоКГ): органічної патології не виявлено.
Консультації фахівців:
- онколог: онкопатології на момент огляду не виявлено;
- гематолог: залізодефіцитна анемія системного генезу;
- алерголог: хронічна кропив’янка;
- генетик: генетичних відхилень не виявлено;
- ендокринолог: аліментарно-конституційне ожиріння;
- імунолог: СШ?
Під час перебування у відділенні проводилася терапія НПЗП, внутрішньовенно глюкокортикоїдами, антигістамінними препаратами.
У зв’язку з тим, що симптомокомплекс у вигляді 1) підвищення запальних маркерів крові, 2) висипу (за типом уртикарного), 3) скелетно-м’язових змін як неприємних відчуттів у м’язах, суглобах та кістках відповідав як критеріям CAPS (кріопіринасоційованому періодичному синдрому), так і вірогідному діагнозу СШ, було прийнято рішення дообстежити дитину методом імуноелектрофорезу фракцій білків крові з імунофіксацією.
Метод імунофіксації підтвердив наявність парапротеїнемії у вигляді моноклонального клону IgG, легкого ланцюга каппа.
На підставі того, що при CAPS-синдромі парапротеїнемія не відмічається [38], у липні 2021 р.на підставі Страсбурзьких критеріїв встановлений вірогідний діагноз: СШ, IgG-асоційований.
Для точного встановлення діагнозу, враховуючи, що у пацієнтки виявлений саме IgG-асоційований варіант моноклональної гамапатії, була необхідність у ще одному критерії, оскільки при даному варіанті захворювання необхідно мати 2 обов’язкових та 3 малих критерії.
У якості додаткового методу дослідження обрана сцинтиграфія, оскільки, як зазначалося вище, саме цей метод має дуже високу чутливість для виявлення кісткових змін при СШ.
При сцинтиграфії виявлено вогнище гіперфіксації остеотропного радіофармпрепарату у 2-му ребрі зліва попереду до 247%. (рис. 4).
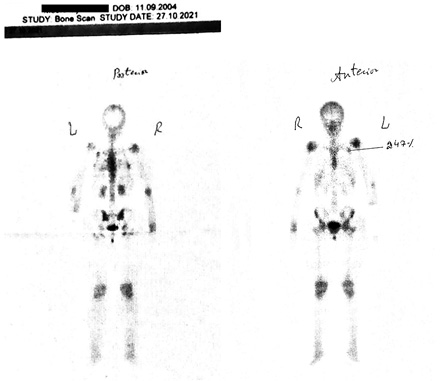
На підставі наявності 2 обов’язкових критеріїв (хронічний уртикарний висип + моноклональна гамапатія IgG) та 3 додаткових критеріїв (рекурентна лихоманка, об’єктивні ознаки аномального ремоделювання кісток з осалгіями, що виявлено за допомогою сцинтиграфії, а також наявному лейкоцитозу та підвищенню СРБ) встановлений заключний діагноз: СШ, IgG-асоційований.
Згідно з рекомендаціями дитині призначено специфічну терапію анакінрою (інгібітором IL-1) [2, 3, 6, 39]. У січні 2022 р. за рахунок державних публічних закупівель лікарнею препарат отриманий, дитина розпочала планову терапію анакінрою у дозуванні 100 мг щоденно у вигляді підшкірних ін’єкцій.
На фоні прийому препарату відмічалося значне покращення загального самопочуття: вже протягом першого тижня у дитини зменшився висип до майже повного зникнення, знизилася інтенсивність неприємних відчуттів у суглобах, кістках, покращилися лабораторні показники (табл. 3).
Таблиця 3. Лабораторні показники пацієнтки Д. на фоні терапії препаратом анакінра
| 20.01.22 (за день до початку прийому анакінри) | 25.01.22 (4 дні після початку прийому) | 19.04.22 (3 міс після початку прийому) | |
|---|---|---|---|
| ШОЕ, мм/хв | 50 | 36 | 6 |
| СРБ, мг/мл | 96 | Негативний | Негативний |
Слід зазначити, що у пацієнтки відмічалися побічні реакції на фоні терапії:
- місцево — почервоніння, ущільнення у місці введення, більше було виражене протягом перших 3 міс лікування;
- системно — дворазово протягом 1 року лікування — прояви у вигляді ларингоспазму, що супроводжувалися вираженою кропив’янкою та купірувалися однією дозою антигістамінного препарату в домашніх умовах.
У подальшому протягом 2022 р. пацієнтка неодноразово проходила обстеження, патологічних змін клінічно та в лабораторних показниках не відмічалося.
У вересні 2022 р. пацієнтка після досягнення віку 18 років переведена на спостереження дорослого ревматолога та сімейного лікаря.
На початок січня 2023 р. (рік з моменту початку терапії інгібітором IL-1) дитина почувалася задовільно, не відмічено висипу, болю у суглобах/кістках/м’язах, лабораторні маркери запалення (ШОЕ та СРБ) були в межах норми.
На жаль, з 20.01.2023 р. пацієнтка препарату не отримує у зв’язку з його відсутністю. 21.01.2023 р. стан дитини погіршився, з’явилися осалгії, уртикарний висип на тілі, свербіж.
Висновки
СШ є рідкісним і недостатньо діагностованим розладом. Висока настороженість щодо СШ має бути при виявленні пацієнтів із хронічними дерматозами, подібними до кропив’янки, періодичною лихоманкою та артралгіями. Таким пацієнтам необхідно проводити дообстеження у вигляді електрофорезу сироваткових білків з імунофіксацією.
Диференційний діагноз має проводитися з:
1) іншими імунологічними захворюваннями, такими як ревматоїдний артрит з системним початком, системний червоний вовчак;
2) гематологічними захворюваннями, а саме: моноклональна гамапатія невідомого генезу, макроглобулінемія Вальденстрема, інші лімфоми, мієломна хвороба;
3) аутозапальними синдромами (такими як кріопіринасоційований періодичний синдром — CAPS);
4) інфекційними захворюваннями, що можуть супроводжуватися висипом та лімфаденопатією, та інші.
Страсбурзькі критерії є основним інструментом для встановлення діагнозу СШ.
Діагноз важливо розпізнати, оскільки СШ пов’язаний із злоякісним новоутворенням. Лімфопроліферативний розлад розвивається у близько 20% пацієнтів в середньому через 7,6 року після появи симптомів. Саме тому пацієнти потребують постійного та тривалого спостереження гематологом для своєчасного виявлення злоякісних новоутворень.
Інгібітори IL-1 вважаються терапією першої лінії, основним завданням яких є пригнічення запалення, проте, на жаль, слід зазначити, що на фоні припинення лікування відмічалося повернення симптомів, тож пацієнти мають отримувати препарат протягом усього життя.
Список використаної літератури
- 1. Jain T., Offord C.P., Kyle R.A. et al. (2013) Schnitzler syndrome: an under-diagnosed clinical entity. Haematologica, 98: 1581–1585. doi: 10.3324/haematol.2013.084830.
- 2. Benitez Gutierrez L.M.P. S, Tutor de Ureta P., Yebra Bango M. (2013) Schnitzler’s syndrome: a case report and literature review of the response to treatment with anakinra. Med. Clin. (Barc), 140: 427–428. 10.1016/j.medcli.2012.09.003.
- 3. Billey T., Beldjerd M., Popa L. et al. (2010) Schnitzler syndrome: a dramatic improvement with anakinra. Presse Med., 39: 1338–1339. 10.1016/j.lpm.2010.07.009.
- 4. Lipsker D. (2010) The Schnitzler syndrome rewiew. Lipsker Orphanet Journal of Rare Diseases, 5: 38. 10.1186/1750-1172-5-38.
- 5. Schnitzler L. (1972) Lésions urticariennes chroniques permanentes (érythème pétaloïde?). J. Dermatol. Angers. 10.1186/1750-1172-5-38.
- 6. de Koning H.D. (2014) Schnitzler’s syndrome: lessons from 281 cases. Clin. Transl. Allergy, Dec 5; 4: 41. 10.1186/2045-7022-4-41.
- 7. de Koning H.D., Bodar E.J., van der Meer J.W., Simon A, Schnitzler Syndrome. Study Group (2007) Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin. Arthritis Rheum., 37(3): 137–48. 10.1016/j.semarthrit.2007.04.001.
- 8. Manthiram K., Zhou Q., Aksentijevich I. et al. (2017) The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat. Immunol.; 18(8): 832–842. 10.1038/ni.3777.
- 9. Cong-Qiu Chu (2022) Schnitzler syndrome and Schnitzler-like syndromes. Chin. Med. J. (Engl.), May 20; 135(10): 1190–1202. 10.1097/CM9.0000000000002015.
- 10. Migliorini P., Italiani P., Pratesi F. et al. (2019) Cytokines and soluble receptors of the interleukin-1 family in Schnitzler syndrome. Scand. J. Rheumatol., May; 48(3): 235–238. 10.1080/03009742.2018.1550210.
- 11. Pathak S., Rowczenio D.M., Owen R.G. et al. (2019)Exploratory study of MYD88 L265P, rare NLRP3 variants, and clonal hematopoiesis prevalence in patients with Schnitzler syndrome. Arthritis Rheumatol.; 71: 2121–2125. 10.1002/art.41030.
- 12. Rowczenio D.M., Pathak S., Arostegui J.I. et al. (2018)Molecular genetic investigation, clinical features, and response totreatment in 21 patients with Schnitzler syndrome. Blood; 131: 974–981. 10.1182/ blood-2017-10-810366.
- 13. Bonnekoh H., Scheffel J., Maurer M. et al. (2018) Use of skin biomarker profiles to distinguish Schnitzler syndrome from chronic spontaneous urticaria: results of a pilot study. Br. J. Dermatol.; 178: 561–562. 10.1111/bjd.15705.
- 14. Takimoto-Ito R., Kambe N., Kogame T. et al. (2021) Refractory serum immunoglobulin M elevation during anti-interleukin (IL)-1- or IL-6-targeted treatment in four patients with Schnitzler syndrome. J. Dermatol.; 48: 1789– 1792. 10.1111/1346-8138.16124.
- 15. Kinoshita M., Shinomiya N., Ono S. et al. (2006) Restoration of natural IgM production from liver B cells by exogenous IL-18 improves the survival of burn-injured mice infected with Pseudomonas aeruginosa. J. Immunol.; 177: 4627–4635. 10.4049/jimmunol.177.7. 4627.
- 16. Airoldi I., Raffaghello L., Cocco C. et al. (2004) Heterogeneous expression of interleukin-18 and its receptor in B-cell lymphoproliferative disorders deriving from naive, germinal center, and memory B lymphocytes. Clin. Cancer Res.; 10: 144–154. 10.1158/1078-0432.CCR-1026-3.
- 17. Kieffer C., Cribier B., Lipsker D. (2009) Neutrophilic urticarial dermatosis: a variant of neutrophilic urticaria strongly associated with systemic disease. Report of 9 new cases and review of the literature. Medicine (Baltimore), 88(1): 23-31. 10.1097/MD.0b013e3181943f5e.
- 18. de Castro F.R., Masouyé I., Winkelmann R.K. et al. (1996) Urticarial pathology in Schnitzler’s (hyper-IgM) syndrome. Dermatology, 193(2): 94–9. 10.1159/000246220.
- 19. Soubrier M., Dubost J.J., Jouanel P. et al. (1994) Multiples complications d’une IgM monoclonale. Rev. Med. Interne., 15: 484–6. 10.1186/1750-1172-5-38.
- 20. Lipsker D., Veran Y., Grunenberger F. et al. (2001) The Schnitzler syndrome. Four new cases and review of the literature. Medicine (Baltimore). Jan; 80(1): 37–44. 10.1097/00005792-200101000-00004.
- 21. Berdy S.S., Bloch K.J. (1991) Schnitzler’s syndrome: a broader clinical spectrum. J. Allergy Clin. Immunol., 87: 849–54. 10.1016/0091-6749(91)90132-8.
- 22. Borradori L., Rybojad M., Puissant A. et al. (1990) Urticarial vasculitis associated with a monoclonal IgM gammopathy: Schnitzler’s syndrome. Br. J. Dermatol., 123: 113–8. 10.1111/j.1365-2133.1990.tb01831.x.
- 23. Lipsker D., Spehner D., Drillien R. et al. (2000) Schnitzler syndrome: heteregous immunopathologic findings involving IgM-skin interactions. Br. J. Dermatol., 142: 954–9. 10.1186/1750-1172-5-38.
- 24. Besada E., Nossent H. (2010) Dramatic response to IL1-RA treatment in longstanding multidrug resistant Schnitzler’s syndrome: a case report and literature review. Clin. Rheumatol., 29: 567–71. 10.1007/s10067-010-1375-9.
- 25. Lecompte M., Blais G., Bisson G. et al. (1998) Schnitzler’s syndrome. Skeletal. radiol., 27: 294–6. 10.1007/s002560050385.
- 26. De Waele S., Lecouvet F.E., Malghem J. et al. (2000) Schnitzler’s syndrome: an unusual cause of bone pain with suggestive imaging features. AJR, 175: 1325–1327. 10.2214/ajr.175.5.1751325.
- 27. Niederhauser B.D., Dingli D., Kyle R.A. et al. (2014) Imaging findings in 22 cases of Schnitzler syndrome: characteristic para-articular osteosclerosis, and the «hot knees» sign differential diagnosis. Skeletal. Radiol., 43: 905–915. 10.1007/s00256-014-1857-y.
- 28. Lebbe C., Rybojad M., Klein F. et al. (1994) Schnitzler’s syndrome associated with sensorimotor neuropathy. J. Am. Acad. Dermatol., 30: 316–318. 10.1016/s0190-9622(94)70031-1.
- 29. Simon A., Asli B., Braun-Falco M. et al. (2013) Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy: European Journal of Allergy and Clinical Immunology. 09 March. 10.1111/all.12129.
- 30. Gladue H.S., Fox D.A., Lowe L. et al. (2014) Schnitzler’s syndrome in the absence of a monoclonal gammopathy: a report of two cases. J. Clin. Cell. Immunol.; 5: 5. 10.4172/2155- 9899.1000265.
- 31. Henning M.A.S., Jemec G.B.E., Ibler K.S. (2020) Incomplete Schnitzler syndrome. Acta Dermatovenerol. Croat.; 28: 38–40. PMID: 32650850.
- 32. Mulla E., Neame R. (2015) Delayed development of the IgM paraprotein in Schnitzler’s syndrome. Scand. J. Rheumatol.; 44: 521–522. 10.3109/03009742.2015.1071421.
- 33. Neel A., Henry B., Barbarot S. et al. (2014) Long-term effectiveness and safety ofinterleukin-1 receptor antagonist (anakinra) in Schnitzler’s syndrome: a French multicenter study. Autoimmun. Rev.; 13: 1035–1041. 10.1016/j. autrev.2014. 08.031.
- 34. Gilson M., Abad S., Larroche C. et al. (2007) Treatment of Schnitzler syndrome with anakinra. Clin. Exp. Rheumatol., 25: 931. PMID: 18173934.
- 35. Perrin F., Neel A., Graveleau J. et al. (2014) Two cases of anakinra-induced neutropenia during auto-inflammatory diseases: drug reintroduction can be successful. Presse Med., 43: 319–321 10.1016/j.lpm.2013.06.028.
- 36. de Koning H.D., Schalkwijk J., van der Ven-Jongekrijg J. et al. (2012) Sustained efficacy of the monoclonal anti-interleukin-1 beta antibody canakinumab in a 9-month trial in schnitzler’s syndrome. Ann. Rheum. Dis. 10.1136/annrheumdis-2012-202192.
- 37. Asli B., Brouet J.C., Fermand J.P. (2011) Spontaneous remission of Schnitzler syndrome. Ann. Allergy Asthma Immunol., 107: 87–88. 10.1016/j.anai.2011.04.006.
- 38 Rowczenio M. et al. (2017) Late-Onset Cryopyrin-Associated Periodic Syndromes Caused by Somatic NLRP3 Mosaicism—UK Single Center Experience. Front Immunol.; 8: 1410. 2017 Oct 31. 10.3389/fimmu.2017.01410.
- 39. Crouch R., Akhras V., Sarkany R. (2007) Schnitzler’s syndrome: successful treatment with anakinra. Australas J. Dermatol., 48: 178–181. 10.1111/j.1440-0960.2007.00375.x.
Адреса для листування:
Мірошниченко Михайло Володимирович
E-mail: kirdol66@gmail.com
Leave a comment