КЛІНІЧНИЙ ВИПАДОК ШВИДКОПРОГРЕСУЮЧОГО УСКЛАДНЕНОГО СЕПСИСОМ ЮВЕНІЛЬНОГО ДЕРМАТОМІОЗИТУ
Ошлянська О.А.1,2, Степанюк Я.С.3, Руденко С.М.3, Лейбюк Ю.В.4, Шатрова К.М.1, Савостікова Н.Л.3, Слободян С.В.3, Синоверська О.Б.4,5
- 1Національний університет охорони здоров’я України імені П.Л. Шупика
- 2Державна установа «Всеукраїнський центр материнства та дитинства НАМН України»
- 3Національна дитяча спеціалізована лікарня «Охматдит» МОЗ України
- 4КНП «Івано-Франківська обласна дитяча клінічна лікарня ІФОР
- 5Івано-Франківський національний медичний університет
Резюме. У статті представлено випадок швидкопрогресуючого ювенільного дерматоміозиту у підлітка, що ускладнився сепсисом та перфорацією дванадцятипалої кишки. Попри застосування сучасних методів лікування, включаючи імунобіологічну терапію, летальність серед таких пацієнтів залишається досить високою (близько 3%). Діагностичні труднощі, тяжкий міопатичний синдром в дебюті захворювання, переривання лікування та розвиток вторинного імунодефіциту на тлі анти-В-лімфоцитарної терапії, стали ключовими факторами несприятливого прогнозу. Висвітлено типові помилки та проблеми ведення пацієнтів із тяжким перебігом ювенільного дерматоміозиту, підкреслено важливість моніторингу імунного статусу та безперервного контролю лікування на всіх етапах ведення таких хворих.
DOI: 10.32471/rheumatology.2707-6970.19582
УДК: 616.5-002+616.74-002]-053.6-06:616.94
Ювенільний дерматоміозит (ЮДМ) — це системне аутоімунне захворювання з групи ювенільних ідіопатичних запальних міопатій, що супроводжується симетричним ураженням проксимальних м’язів, характерним ураженням шкіри та васкулопатією. ЮДМ є найпоширенішою формою ювенільних ідіопатичних запальних міопатій, і разом з тим — орфанною патологією із захворюваністю 1:250 000–500 000 дітей. Середній вік дебюту 5–14 років, при цьому дівчата хворіють частіше, ніж хлопці (2–5:1) [5].
Доведено, що в патогенезі ЮДМ важливе значення відіграють генетичні фактори: виявлені асоціації з алелями HLA-DQA1*0501, HLA-DQA*0301, HLA-DRB1*0301 генів головного комплексу гістосумісності, а також мутації в певних генах, що кодують промотор фактора некрозу пухлин-α, інтерлейкін-1β, антагоніст рецептора інтерлейкіну-1 інтрон, регуляторний фактор інтерферону 5, манозозв’язувальний лектин, хемокіновий ліганд 21, подібний до фосфоліпази С білок тощо, які призводять до розвитку генетично-асоційованих варіантів ЮДМ [3]. Епігенетичні чинники, такі як інфекційні агенти (віруси Коксакі, грипу, гепатиту В, парвовірус, стрептококи групи А, токсоплазми та борелії), вакцини, ультрафіолетове випромінювання та деякі лікарські засоби, можуть виступати вагомими тригерами розвитку ЮДМ [3].
Діагноз ЮДМ встановлюється на підставі класифікаційних критеріїв Європейського альянсу ревматологічних асоціацій (European Alliance of Associations for Rheumatology — EULAR) / Американського коледжу ревматологів (American College of Rheumatology — ACR) (2017), які можуть використовуватися як з урахуванням результатів біопсії м’язів, так і без цього, і розцінюються як більш специфічні порівняно з критеріями за Bohan and Peter (1975), особливо за наявності результатів біопсії м’язів [7]. Диференційний діагноз в дитячому та підлітковому віці передбачає виключення м’язової дистрофії, метаболічних захворювань, вірусних міозитів тощо. При встановленні діагнозу ЮДМ усім пацієнтам необхідно проводити скринінг на інтерстиціальне ураження легень за допомогою комп’ютерної томографії та деяких біомаркерів у сироватці крові (наприклад білок Кребс фон ден Лунген-6) [3]. Це є вкрай необхідним, оскільки пацієнти з інтерстиціальним ураженням легень, тяжкою міопатією (до знерухомлення), генералізованим набряком, дисфагією, васкулопатією шлунково-кишкового тракту (ШКТ) (сильний біль та кровотеча), ураженням серця та центральної нервової системи потребують більш агресивного лікування [2]. Терапія системними глюкокортикостероїдами (ГКС) і досі залишається основою лікування ЮДМ. Раннє та агресивне лікування високими дозами ГКС (включаючи пульс-терапію) у поєднанні з іншими імуносупресантами (метотрексат, циклофосфамід, циклоспорин А, мікофенолату мофетил) застосовується також при тяжких та фульмінантних випадках. У пацієнтів із рефрактерним ЮДМ додатково використовується терапія високими (супресорними) дозами внутрішньовенного імуноглобуліну та призначаються імунобіологічні засоби, переважно анти-В-лімфоцитарні препарати (ритуксимаб), інгібітори фактора некрозу пухлин, інгібітори янус-кіназ. Останнім надається перевага у випадках з наявністю «генетичних» ЮДМ [3, 8]. Останніми роками для лікування хворих на ЮДМ пропонується аутологічна таргетна CAR T-клітинна терапія, при якій пацієнту вводяться власні модифіковані Т-лімфоцити, які є тропними до власних В-лімфоцитів [6].
Проте незважаючи на всі сучасні досягнення у лікуванні, летальність хворих на ЮДМ залишається досить високою та становить близько 3% випадків [12]. Сьогодні з’ясовано, що несприятливий перебіг захворювання та зростання ризику смертності у пацієнтів з ЮДМ зумовлюють затримка встановлення діагнозу та відстрочений початок лікування, погана прихильність до лікування, виражений міопатичний синдром під час маніфестації хвороби, повторні інфекційні захворювання в перші роки від дебюту захворювання [12]. Найчастішими безпосередніми причинами смерті при ЮДМ є васкулопатичні ускладнення з боку ШКТ (при перфорації ШКТ 33,3–38,0% випадків летальні [13, 14]) та ураження легень (28,9% усієї летальності протягом 2 міс від появи респіраторних симптомів [11]). До менш частих причин смерті при ЮДМ належать прогресуючий міокардит (14,0–55,0% летальних випадків [10]) та інтеркурентні інфекції (сепсис на тлі імуносупресивної терапії становить близько 1% в структурі летальності при ЮДМ) [12].
За даними попередніх досліджень, виявлено декілька факторів високого ризику смертності від ускладнень з боку ШКТ: тяжкий міопатичний та шкірний синдром в дебюті (зі шкірними виразками), низький (<15) індекс маси тіла, позитивні антинуклеарні та анти-NXP2-антитіла [13]. Найчастіше перфорація відбувається у дванадцятипалій кишці в перші 10 міс після встановлення діагнозу у пацієнтів, які з приводу високої активності захворювання отримують високодозову ГКС-терапію [13, 14]. Першими симптомами перфорації ШКТ при ЮДМ є прогресуючий біль у животі та підвищення температури тіла [13].
З метою аналізу типових помилок тактики ведення та лікування наводимо спостереження випадку тяжкого швидкопрогресуючого ЮДМ у підлітка, що ускладнився сепсисом та перфорацією ШКТ.
На опис клінічного випадку отримано інформовану згоду батьків пацієнта.
Хлопчик А., 15 років. З анамнезу життя відомо, що дитина від ІІ вагітності, народжений від неспорідненого шлюбу. У родині сибси (старша сестра та молодший брат) пацієнта здорові. Фізичний розвиток відповідав віковій нормі. До актуального епізоду серйозних та хронічних соматичних захворювань не мав. Щеплений відповідно до календаря вакцинації. З 5-річного віку спостерігався неврологом з приводу епілепсії з атонічними нападами. В анамнезі прийом кількох протисудомних препаратів, на час актуального звернення приймав депакін та ламотриджин. Перебував на диспансерному обліку у психіатра з приводу затримки психічного розвитку. Навчався інклюзивно. Навички самообслуговування розвинуті.
Захворів гостро, коли на 3-тю добу після екстракції зуба батьки відмітили утворення набряку під правим оком та появу висипу в ділянці перенісся. За місцем проживання вказані симптоми були розцінені як інфекційно-алергічна реакція. Призначення антибіотикотерапії (цефтріаксон внутрішньовенно протягом 5 днів) та одноразове введення дексаметазону успіху не мало. Приєднався біль в ділянці грудної клітки та в нижній частині спини, стала затрудненою хода. 26.07.2024 р. дитина госпіталізована в Обласну дитячу клінічну лікарню (ОДКЛ) за місцем проживання (рис. 1).
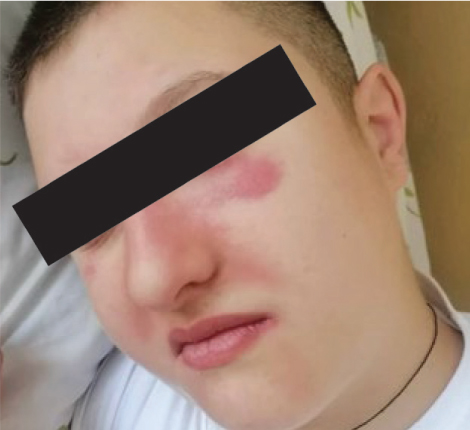
Загальний стан при госпіталізації розцінювався як середньої тяжкості, за захворюванням — ближче до тяжкого. Дитина оглянута консилярно педіатром, кардіоревматологом, дерматологом, алергологом. Наявна клінічна симптоматика потребувала дообстеження з метою виключення інсектної алергії, паранеопластичного синдрому, постковідного синдрому та гострої запальної міопатії. У приймальному відділенні ОДКЛ зафіксовано генералізований напад судом, з приводу якого пацієнта було госпіталізовано у відділення анестезіології та інтенсивної терапії. Через 2 дні переведений у реабілітаційне відділення. Під час обстеження в загальному аналізі крові відмічений помірний лейкоцитоз (10,7 Г/л), швидкість осідання еритроцитів (ШОЕ) 10 мм/год. У біохімічному аналізі — підвищення рівня трансаміназ (аланінамінотрансфераза (АлАТ) втричі вище за нормальні значення, аспартатамінотрансфераза (АсАТ) — в 10 разів більше), активність креатинфосфокінази (КФК-МВ) — 288 Од/л (норма до 25 Од/л), вміст С-реактивного білка (СРБ) — 12 мг/л (норма до 6 мг/л), підвищення рівня Д-димеру до 1190 нг/мл (норма до 500 нг/мл), показники коагулограми в межах норми. Проведена рентгенографія органів грудної клітки змін не виявила. Продовжено курс антибіотикотерапії (цефазолін із амікацином), призначено інфузійну терапію. Протягом наступних 2 діб відмічалася поява вираженої загальної слабкості, болю у м’язах, захитування при ходьбі, підвищення температури тіла до субфебрильних цифр. З 30.07.2024 р. відмічені збільшення набряку обличчя, поява набряку м’яких тканин шиї (рис. 2).
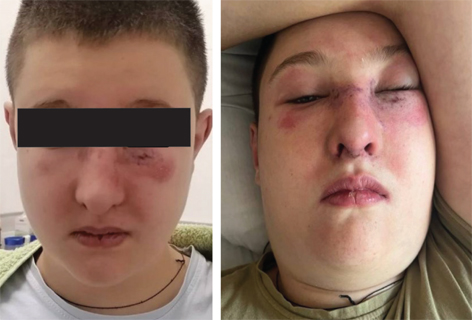
За даними комп’ютерної томографії (КТ) головного мозку, органів грудної клітки, черевної порожнини та малого таза з внутрішньовенним контрастуванням від 31.07.2024 р., ознак неопластичного процесу не виявлено. За результатами ультразвукового дослідження (УЗД) серця — гідроперикард (перикардіальний випіт по задній стінці до 12 мм, по передній — 3 мм, на верхівці — 6 мм).
З 02.08.2024 р. — збільшення вираженості міалгії та м’язової слабкості: хлопчик не міг самостійно підняти голову, руки та ноги, повернутися в ліжку. Об’єктивно: геліотропний висип і збільшення набряку параорбітально, поява папул Готтрона, сітчасте ліведо на кінцівках та тулубі, афтозний стоматит та гінгівіт. Пальпаторно м’язи кінцівок щільні та болючі. Параклінічно: підвищення активності КФК (до 17 310 Од/л), рівня АсАТ (до 527 Од/л), АлАТ (до 106 Од/л), лактатдегідрогенази (ЛДГ) до 839 Од/л. На підставі клінічних та лабораторних даних дитині встановлений діагноз ЮДМ.
Враховуючи тяжкість стану та швидкість збільшення вираженості симптоматики, 5–7.08.2024 та 12–14.08.2024 р. проведена комбінована пульс-терапія (1000 мг метилпреднізолону та 1000 мг циклофосфаміду), введено прийом преднізолону 1,5 мг/кг/добу перорально. Стан дитини в динаміці з незначним покращенням: зменшилися набряк м’язів, еритема шкіри, незначно зросла рухова активність.
З 27.08.2024 р. стан хворого повторно погіршився: поява фебрильної лихоманки, задишки, різке пригнічення ковтального та кашльового рефлексів. 29.08.2024 р. у зв’язку зі збільшенням вираженості проявів десатурації переведений у відділення інтенсивної терапії. Введено внутрішньовенно імуноглобулін в дозі 0,8 г/кг маси тіла. Стан без суттєвої позитивної динаміки. Враховуючи тяжкий швидко прогресуючий, рефрактерний до терапії ГКС та цитостатиками перебіг захворювання, ухвалено консилярне рішення про інтенсифікацію терапії за життєвими показаннями. 05.09.2024 р. пацієнту введено ритуксимаб в дозі 1000 мг. Стан пацієнта залишався тяжким за рахунок вираженого міастенічного синдрому, дихальної недостатності тяжкого ступеня, інтоксикаційного синдрому. Безперервно проводилася апаратна та бронхоскопічна санація бронхоальвеолярного апарату. Годування проводилося через назогастральний зонд.
11.09.2024 р. за наполяганням батьків переведений в НДСЛ «ОХМАТДИТ» (м. Київ) у відділення інтенсивної терапії. При надходженні загальний стан дитини розцінено як вкрай тяжкий через дихальну недостатність та виражений міопатичний синдром. Самостійні рухи відсутні, відмічалася мінімальна рухова активність в дистальних відділах кінцівок. Самостійно не кашляв, не ковтав. На шкірі — сітчасте ліведо, набряк під правим оком. На слизовій оболонці рота — множинні афти, гінгівіт, хейліт. Дихання спонтанне за участю допоміжних м’язів з подачею кисню 14 л/хв через маску, сатурація кисню (SpO2) — 69–79%. Відмічалися приглушеність серцевих тонів, тахікардія 130–150 уд./хв, вислуховувався систолічний шум на верхівці серця, артеріальний тиск (АТ) — 130–140/75–80 мм рт. ст. Живіт доступний для глибокої пальпації, печінка та селезінка не виступали з-під країв реберних дуг.
Відсутність типових шкірних проявів на момент огляду у відділенні інтенсивної терапії призвела до думки про неточність попереднього діагнозу ЮДМ. У плані диференційної діагностики розглядалися вроджені міопатії та мітохондріальні захворювання. На час проведення диференційної діагностики було тимчасово відмінено системну ГКС-терапію. У відділенні інтенсивної терапії пацієнт отримував антибактеріальну, протигрибкову терапію, гемотрансфузії за показаннями.
Параклінічно в динаміці: підвищення лейкоцитозу (до 14 Г/л), поява анемії (гемоглобін 98 г/л), та підвищення ШОЕ (30 мм/год) на тлі тенденції до нормалізації попередньо виявлених біохімічних відхилень (АлАТ 29 Од/л, АсАТ 36 Од/л, ЛДГ 372 Од/л, КФК 337 д/л, феритин 955,7 мкг/л, СРБ 37,9 мг/л). Імунограма: імуноглобулін G — 8,85 г/л, IgA — 1,90 г/л, IgM — 1,21 г/л. Імуноцитофлуориметричне дослідження: відсутність В-лімфоцитарної субпопуляції (абсолютна та відносна кількість В-лімфоцитів дорівнювали 0,0), звуження субпопуляції Т-лімфоцитів (0,27·109/л при нормі 0,44–2,74), Т-лімфоцитів-хелперів (0,10·109/л при нормі 0,27–1,98), знижений імунорегуляторний індекс (до 0,55).
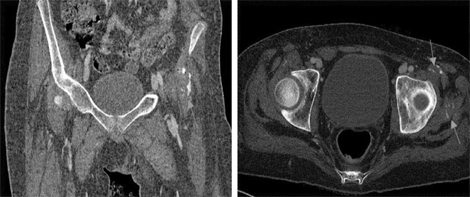
За даними мультиспіральної КТ: субтотальна консолідація паренхіми нижньої частки лівої легені із наявністю «повітряної бронхографії», на окремих рівнях просвіти бронхів місцями не простежувалися; в базальних відділах правої легені виявлені паренхіматозні фіброзні тяжі, в паренхімі обох легень ділянки ущільнення за типом «матового скла». КТ м’язів: гіпотрофія з ділянками набряково-інфільтративного характеру. У сідничних та поперекових м’язах — ділянки нерівномірної кальцифікації (рис. 3).
11.09.2024 р. у зв’язку із прогресуючою дихальною недостатністю хлопчика переведено на вентиляцію легень через інтубаційну трубку; санувалася велика кількість гнійного мокротиння. З 12.09.2024 р. ГКС відмінені. 26.09.2024 р. через неефективність самостійного дихання накладено трахеостому, через яку продовжувалася штучна вентиляція легень. 30.09.2024 р. відновлене самостійне дихання з подачею кисню 3–4 л/хв, після чого дитина ще у тяжкому стані переведена до педіатричного відділення.
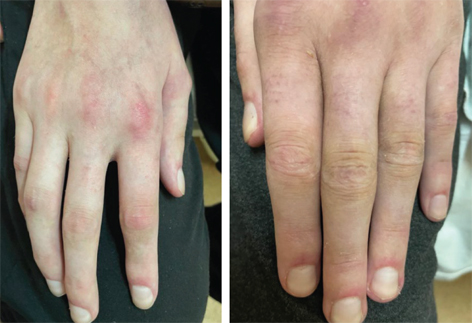
На момент переведення утримувалася дихальна недостатність І ст. (частота дихання 24/хв, SpO2 97%), частота серцевих скорочень 125 уд./хв, АТ 123/63 мм рт. ст., повна знерухомленість, атрофія м’язів, афонія, мінімальна еритема на обличчі, бляшки Готтрона, почервоніння навколонігтьових валиків (рис. 4), сформувалися трофічні порушення в ділянці куприка, відмічені позиційні периферичні набряки кінцівок, поява контрактур в колінних, ліктьових, променево-зап’ясткових суглобах та дрібних суглобах кистей. Харчування було зондовим, дихання самостійне через трахеостомічну трубку.
Параклінічно в динаміці: виявлені імуноглобуліни класу G до S1-білка SARS-CoV-2 (1776,1 ОЕ/мл) та SARS-CoV-2-IgG (252,2 BAU/мл), антитіла до двоспіральної ДНК негативні, виявлені пограничні значення антитіл міозитного профілю — анти-MI-2 та анти-тРНК-антитіл (незважаючи на попередню агресивну ГКС та імуносупресивну терапію).
Враховуючи затримку статевого розвитку, визначені рівні статевих гормонів (тестостерон <0,025 нг/мл, пролактин 29,19 нг/мл, фолікулостимулючий гормон 0,445 мМОд/мл, лютеїнізуючий гормон <0,1 мМОд/мл), хлопчик консультований ендокринологом (правобічний паховий крипторхізм, вторинний гіпогонадизм).
За даними магнітно-резонансної томографії (МРТ) головного мозку та шиї: зменшення об’єму великих півкуль та мозочка, вентрикулодилатація без ознак порушення ліквородинаміки, арахноїдальні кісти задньої черепної ямки, дифузний набряк м’язів спини та шиї. Голкова електронейроміографія: наявність міопатичного типу ураження дифузно (низькоамплітудні потенціали, менше 100 мкВ (при нормальних значеннях 300–600 мкВ), значно укорочені до 5 мс, поліфазні), відмічена нейропатія малогомілкових нервів та радикулопатія крижових відділів.
Мітохондріальний геном був оцінений методом секвенування нового покоління: мутацій не виявлено. Каріотип: 46, XY. Виявлені мутації в генах фолатного обміну (алельний поліморфізм 677СТ та 1298АС гена MTHFR в гетерозиготному стані).
Після 1 міс відміни ГКС-терапії в динаміці параклінічно підвищення тромбоцитозу (до 593 Г/л), ШОЕ (до 74 мм/год), феритину (до 2775,3 мкг/л), при цьому КФК (254 Од/л) та СРБ (6,3 мг/л) були підвищені незначно.
Враховуючи наявність у дитини гострого тяжкого міопатичного синдрому (тотальна м’язова слабкість до знерухомлення, міалгії, опис стану м’язів за оглядом в ОДКЛ, відсутність повідомлення про наявність м’язових порушень з раннього віку, синдром міолізу з максимальними значеннями КФК 17 310 МО/л з подальшою позитивною динамікою на тлі лікування (ГКС, цитостатики, довенний імуноглобулін, ритуксимаб), утримання запальних змін з боку скелетних м’язів за даними МРТ та КТ, характерними даними електронейроміографії через 2 міс від початку ГКС-терапії); наявність типових шкірних змін у вигляді периферичних набряків, позитивного симптому Готтрона та геліотропної еритеми з позитивною динамікою на тлі протизапальної терапії; наявність змін з боку слизових оболонок, зафіксованих в медичній документації за місцем проживання; виявлення міозитспецифічних аутоантитіл; залучення ШКТ (дисфагія), серцево-судинної системи (міоперикардит); органів дихання (ураження гортані та ознаки інтерстиціального ураження легень за даними КТ), високої активності запального процесу (підвищені ШОЕ, СРБ, феритин, лейкоцитоз), виключення інфекційного міозиту та злоякісних новоутворень, захворювання дитини розглядалося як ЮДМ (ACR 2017 IIMs=15,4; що навіть без урахування даних біопсії м’язів відповідало достовірному діагнозу) з наявністю вторинного імунодефіцитного стану з порушенням гуморальної ланки імунної відповіді (внаслідок отриманої анти-В-клітинної терапії, що відповідало її ефективності). Супутніми діагнозами визнані затримка психічного, статевого розвитку та неуточнена епілепсія.
Від проведення біопсії м’язів вирішено утриматися, зважаючи на тяжкість стану пацієнта. Враховуючи анамнез (затримка психічного розвитку, епілептичний синдром) та швидко прогресуючий характер ЮДМ, проведено генетичне дослідження (пакет аутозапальних, метаболічних та неврологічних захворювань) в лабораторії INVITAE, США. Виявлено один патогенний варіант у гені CHD2, що асоційований з аутосомно-домінантною епілептичною енцефалопатією, яка починається в дитинстві. У дослідження частково включені гени, які асоційовані з моногенними ЮДМ, мутацій в них не виявлено.
Проведена корекція лікування згідно з рекомендаціями лікування ЮДМ ACR (2017) та Національного інституту здоров’я і досконалості допомоги (National Institute for Health and Care Excellence — NICE) (2022). Відновлено прийом метилпреднізолону в дозі 1 мг/кг маси тіла за преднізолоном, циклофосфамід 100 мг/добу, ніфедипін 30 мг/добу, проводилася кінезіотерапія. Продовжено протисудомну (депакін, ламотриджин), гастропротекторну (омепразол) та антигіпертензивну (еналаприл) терапію. Поступово відмінена антибіотикотерапія.
Враховуючи наявність хронічного аспіраційного синдрому, що персистував у пацієнта на тлі годування через назогастральний зонд та був одним із провокативних факторів дифузного гнійного ендобронхіту, ухвалено рішення про встановлення гастростоми (11.10.2024 р.).
За даними фіброезофагогастродуоденоскопії (ФЕГДС), в препілоричному відділі по передній стінці візуалізована ділянка слизової оболонки діаметром до 2 см з глибокими виразковими вадами (сформована трьома виразками, дно яких вистелено білим цупким фібрином). Додатково до лікування інгібіторами протонної помпи введено блокатори гістамінних рецепторів та репаранти.
У динаміці через 1 міс стан дитини характеризувався вираженою позитивною динамікою міопатичного синдрому. Пацієнт тривало самостійно утримував голову, перевертався з боку на бік, сідав з підтримкою та сидів до 3–4 год на день, вертикалізувався в допоміжному пристрої до 30 хв 2–3 рази на день, піднімав руки до рівня плечей, грав на телефоні, нарізав самостійно фрукти. Відновився голос. Повністю регресували набряки та ущільнення м’язів. Утримувалися періодична еритема в ділянці щік, еритема Готтрона, «мармуровість» шкіри. За даними лабораторного обстеження, зберігалися анемія (96 г/л), гіперферитинемія (734,3 мкг/л), знизилися ШОЕ (до 13 мм/г), АлАТ 32 Од/л, АсАТ 23 Од/л, ЛДГ 237 Од/л, КФК 36 Од/л, СРБ негативний, достатній рівень сироваткових імуноглобулінів (G — 7,38 г/л, A — 1,32 г/л, M — 0,44 г/л) при одночасній відсутності В-лімфоцитів в периферичній крові.
Оскільки при контрольній електроенцефалографії патології не виявлено, була мінімізована протисудомна терапія.
За даними МРТ опорно-рухового апарату в динаміці, відмічений остеонекроз голівок стегнових кісток, атрофічні та запальні зміни м’язів (дифузне підвищення сигналу на Т2-режимі та характерні ознаки перимускулярного набряку, які є типовими для запальних міопатій) (рис. 5).
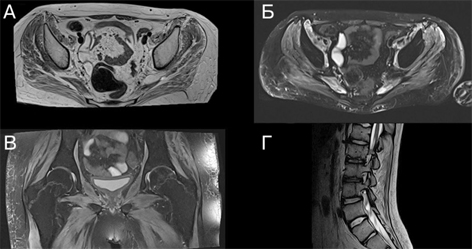
За даними денситометрії — остеопороз (Z-score Total body — –2,4 SD), з приводу цього призначене лікування бісфосфонатами (золендронова кислота).
Оскільки за даними нічної полісомнографії виявлено наявність синдрому апное-гіпопное середнього ступеня тяжкості, пацієнту періодично проводилася неінвазивна вентиляція легень.
При повторному бактеріологічному дослідженні мокротиння виявлено Pseudomonas aeruginosa, тому крім санації інтубаційної трубки додатково застосовували місцеву антибактеріальну терапію (колістин).
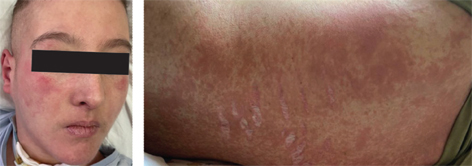
З 05.11.2024 р. відмічалося збільшення вираженості еритеми на обличчі з появою деструктивних ділянок та дисемінованої еритеми на тулубі зі свербежем (рис. 6), незначне підвищення ЛДГ 349 Од/л, з приводу чого до терапії додані хінолінові засоби та топічні препарати такролімусу.
27.11.2024 р. проведено контрольне ендоскопічне дослідження: за даними ФЕГДС навколо гастростоми на слизовій оболонці нашарування гною, слизова оболонка шлунка у всіх відділах гіперемована, набрякла, в препілоричному відділі по передній стінці наявна глибока виразкова вада розміром до 2,5 см, дно якої вистелено білим цупким фібрином, слизова оболонка цибулини дванадцятипалої кишки із множинними виразками розміром до 3 мм. Проведені заміна гастростоми, корекція терапії (зміна інгібітора протонної помпи з омепразолу на езомепразол, додано антибактеріальну терапію (цефтазидим / авібактам). За даними бронхоскопії, зберігався дифузний ерозивно-геморагічний гнійний ендобронхіт. Дитина неодноразово консультована хірургом.
У зв’язку з вираженою імуносупресією за результатами оцінки субпопуляцій лімфоцитів крові та персистенцією інфекційних ускладнень продовження імунобіологічної терапії вирішено відтермінувати.
За наполяганням батьків у стабільному стані 10.12.2024 р. хлопчик виписаний під спостереження ревматолога за місцем проживання. З 11.12.2024 р. дитина перебувала вдома під спостереженням сімейного лікаря.
З 26.12.2024 р. загальний стан дитини різко погіршився: підвищилася температура тіла до субфебрильних цифр, виникли блювання, діарея, біль у животі. Втім по медичну допомогу батьки звернулися аж 30.12.2024 р. За тяжкістю стану госпіталізований у відділення інтенсивної терапії. Проводився динамічний моніторинг ревматологом та хірургами. Стан дитини залишався стабільно тяжким з домінуванням больового абдомінального синдрому, формуванням динамічної кишкової непрохідності. Параклінічно: лейкопенія (2,6 Г/л), підвищення ШОЕ (66 мм/год), гіпопротеїнемія (47 г/л). За даними УЗД та КТ органів черевної порожнини (ОЧП), переконливих даних щодо гострої хірургічної патології не встановлено.
За наполяганням батьків 31.12.2024 р. пацієнт переведений у НДСЛ «ОХМАТДИТ» з діагнозом «Динамічна кишкова непрохідність? Гострий апендицит?». При поступленні 31.12.2024 р. температура тіла 36,8 °С, частота серцевих скорочень 120 уд./хв, АТ 100/70 мм рт. ст., частота дихання 32/хв, SpО2 97% без дотації кисню. При огляді скаржився на біль в ділянці гастростоми та правій здухвинній ділянці. Об’єктивно загальний стан дитини тяжкий за рахунок больового абдомінального синдрому. На обличчі еритема. Дихання через трахеостому ефективне, кількість мокротиння помірна. Аускультативно дихання проводилося над всіма легеневими полями. Живіт помірно піддутий, симетричний, при пальпації болючий в місці виходу гастростоми, набряк і почервоніння шкіри навколо устя гастростоми. У лівій здухвинній ділянці пальпувалися петлі товстого кишечнику, переповнені щільними каловими масами. У правій здухвинній ділянці відмічені біль при глибокій пальпації, слабкопозитивні симптоми подразнення очеревини. У проєкції підребер’я справа — крепітація. Стул самостійний, рідкий, світло-коричневого кольору. Блювання не було.
За даними оглядової рентгенографії ОЧП — підшкірна емфізема; за даними УЗД — вільна рідина в черевній порожнині не візуалізувалася. При референтній оцінці наданої КТ відмічена наявність повітря в заочеревинному просторі, проєкції підшлункової залози, позаду дуоденум та в брижі навколо судин. Червоподібний відросток був у типовому місці та не мав змін.
01.01.2025 р. о 03:00 загальний стан дитини різко погіршився, дитина стала недоступною для продуктивного контакту, агресивною, збудженою, збільшувалася вираженість абдомінального больового синдрому, дихання утруднилося, відмічене зниження сатурації кисню до 90%. На повторній рентгенографії виявлені ателектази легень, під лівим куполом діафрагми — наявність вільного газу в черевній порожнині.
О 12:00 01.01.2025 р. — сопорозний стан, гримаса болю на обличчі, значне збільшення живота, відсутність перистальтики. По стомі у великій кількості відходили зловонні виділення насиченого зеленого кольору. Крепітація (підшкірна) — по всій поверхні живота. Під час транспортування з метою проведення подальшого обстеження відмічена зупинка серцевої діяльності. О 13:00 розпочато серцево-судинну реанімацію, реанімаційні заходи протягом 45 хв виявилися неефективними. О 13:45 констатовано біологічну смерть.
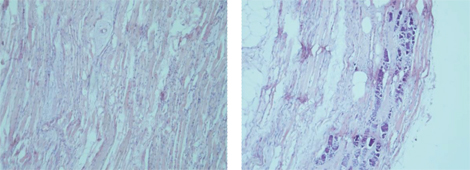
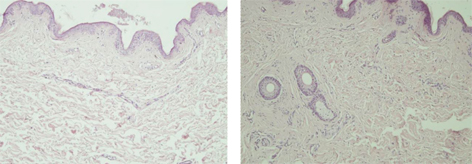
Проведено аутопсію пацієнта А. з клінічним діагнозом ЮДМ. Результати аутопсійного дослідження гістологічних препаратів остаточно підтвердили діагноз дерматоміозиту, який проявлявся не лише тяжким ураженням скелетних м’язів та шкіри (рис. 7, 8), але й множинними ураженнями вісцеральних органів (рис. 9, 10), що свідчило загалом про надвисоку активність аутоімунного запального процесу і, відповідно, про правильне і сучасне обрання тактики лікування.
Зафіксовані також ятрогенні зміни за типом патоморфозу внаслідок вторинного імунодефіциту (делімфатизація селезінки та лімфатичних вузлів — рис. 11), який призвів до розвитку септичного процесу з відсутністю адекватної перифокальної запальної реакції з переважанням некрозів з колоніями бактерій в них (рис. 12). Ознаки імунодефіцитного стану були пов’язані з використанням високодозової імуносупресивної та імунобіологічної терапії препаратами, що безпосередньо пригнічують клітини гуморальної ланки імунної відповіді.
Встановлений заключний патогістологічний діагноз: основне захворювання — ЮДМ з ураженням мʼязів, шкіри, ШКТ, легень. Ускладнення — сепсис (синьогнійно-клебсієльний): некротичний ентероколіт, вогнищева некротична пневмонія з колоніями бактерій. Перфорація стінки дванадцятипалої кишки. Фібринозний перитоніт. Хронічні виразки пілоричного відділу шлунка. Гіпертрофія міокарда лівого шлуночка. Дистрофія міокарда, печінки, нирок. Набухання головного мозку. Безпосередньою причиною смерті пацієнта став синдром поліорганної недостатності.
Обговорення
Аналіз перебігу хвороби в описаному випадку виявив декілька спірних моментів, які призводили до затримки терапевтичних рішень. Першочергово звертало на себе увагу те, що незважаючи на наявність у пацієнта ознак запальної міопатії — гострого тяжкого міопатичного синдрому, відсутності повідомлення про наявність м’язових порушень з раннього віку, синдрому міолізу, виявлення запальних змін з боку скелетних м’язів за даними МРТ та КТ, електронейроміографії, наявності типових шкірних змін та ураження слизових оболонок і внутрішніх органів, виявлення міозитспецифічних аутоантитіл — продовжувався диференційно-діагностичний пошук щодо вроджених міопатичних синдромів, зумовлених вродженими міопатіями, хворобами нервово-м’язового з’єднання чи мітохондріальної патології, які можуть розглядатися у дитини в якості диференційного діагнозу із запальними міопатіями, враховуючи наявність затримки психічного розвитку в анамнезі (таблиця).
| Характеристика | Захворювання | |||
| ЮДМ | Метаболічні міопатії | Мітохондріальні міопатії | Хвороби нервово-м’язового з’єднання | |
| Вік дебюту | 5–14 років, можливі прояви в ранньому дитинстві | Від народження до проявів у дорослому віці | Від народження до проявів у дорослому віці | Від народження до проявів у дорослому віці |
| Шкірні ознаки | Геліотропна еритема, папули / еритема Готтрона, малярний висип, сітчасте ліведо, виразки | Рідко, неспецифічні | Алопеція, гіпертрихоз, акроціаноз, гіпер- чи гіпопігментація | Рідко, не специфічні |
| Ураження м’язів | Проксимальне ураження м’язів переважає над дистальним | Переважання динамічних симптомів над статичними (непереносимість фізичних навантажень при нормальній м’язовій силі).
Можливе проксимальне ураження м’язів або дистальне. |
Дистальне ураження м’язів переважає над проксимальним | |
| Кальцинати | + | – | – | – |
| Ураження легень | + | + | + | – |
| Артрити | + | + | + | – |
| Ураження ССС | + | + | + | + |
| Інші типові прояви | Міалгії, генералізовані набряки, феномен Рейно | Рецидивність, зворотність, декомпенсація після голодування, висококалорійного харчування, холоду чи гострих інфекцій, періодична гіпоглікемія | Низькорослість, туговухість, ретинопатія, ендокринопатія, атаксія, птоз | Виражені деформації хребта |
| Специфічні лабораторні показники | Міозитспецифічні аутоантитіла | Метаболічний ацидоз, низький чи високий рівень карнітину в плазмі крові, дикарбоксильна ацидурія, дефіцити специфічних ферментів (альфа-глюкозидази, фосфофруктокінази і т.ін.) | Лактатацидоз в крові, порушення співвідношення лактат / піруват, високий лактат в лікворі | Немає |
| Біопсія м’язів | Перифасцикулярна атрофія, мікроінфаркти, периваскулярний/ендо-, перимізіальний запальний інфільтрат, облямовані вакуолі | Включення глікогену, ліпідів;
деякі види структурно нормальні |
Рвано-червоні волокна; численні міофібрилярні мітохондрії | Неспецифічні зміни за типом міопатичного патерну |
Примітка: ССС — серцево-судинна система.
Як показано в таблиці, особливості перебігу хвороби у дитини та дані проведеного обстеження одразу свідчили на користь запальної міопатії. Проте підозра на вроджені міопатії призвела до переривання ГКС-терапії, що викликало погіршення подальшого перебігу захворювання, прогресування вісцеральних уражень, потребу встановлення трахеостоми та гастростоми. Останнє зумовило формування додаткових вхідних воріт для інфекційних агентів у імуноскомпрометованого пацієнта.
З коморбідними станами та стійкою імуносупресією пов’язане подальше відтермінування рішення про продовження імунобіологічної терапії. Зазначене потенційно ще більше погіршувало віддалений прогноз перебігу основного захворювання та призвело до прогресування деструктивних змін в тканинах внаслідок персистуючої васкулопатії, що формувало хибне коло незворотних уражень внутрішніх органів.
Описані у хворого клінічні прояви в термінальній стадії хвороби (біль та гіпертермія з попередньо виявленим ураженням ШКТ та наявністю васкуліту за основним діагнозом, почервоніння навколо гастростоми) та інструментальні зміни (наявність повітря у внутрішніх порожнинах) потребували негайної інтенсифікації лікування та проведення лапаротомії з метою ревізії стану ОЧП, яка своєчасно не була виконана.
Необхідно відмітити, що обсяг швидко отриманого генетичного обстеження (гени фолатного обміну) не свідчив на користь будь-яких вроджених міопатій. У подальшому отримані результати більш детального генетичного обстеження, які включали значну кількість генів, що пов’язані з моногенними ЮДМ (а саме гени, які кодують субодиницю А, В та С рибонуклеази Н2, SAM та HD домен, що містить дезоксинуклеозидтрифосфаттрифосфогідролазу 1, три екзонуклеази первинної репарації 1, інтерферонстимульований ген 15, субодиницю рецепторів альфа- та бета-інтерферону 1 та 2, рецептор гамма-інтерферону 1 та 2, інтерфероновий регуляторний фактор 7, Toll-подібний рецептор 7, ламін А/С). Проте в зазначений перелік увійшли не всі відомі сьогодні гени, асоційовані з розвитком ЮДМ. У разі виявлення мутацій в зазначених генах могла розглядатися таргетна терапія інгібіторами янус-кіназ. Очікування результатів генетичного дослідження також зумовило відтермінування терапевтичних рішень.
Узагальнюючи вищезазначене, враховуючи тяжкість загального стану дитини, наявність первинного поліорганного ураження, агресивного перебігу хвороби, летальний випадок був потенційно передбачуваним. Особливостями описаного випадку були швидкість розвитку симптоматики і наявність супутньої генетичної патології. Основний недолік ведення пацієнта — недостатня узгодженість дій мультидисциплінарної команди, недооцінка тяжкості стану пацієнта та обмеження логістичних можливостей в сучасних умовах.
Висновки
ЮДМ є тяжким рідкісним потенційно летальним захворюванням в дитячому віці. Труднощі диференційної діагностики та тимчасове переривання лікування в описаному випадку призвели до прогресування хвороби, необхідності інтенсифікації, подовження тривалості, необхідності застосування високих доз протизапальної та імуносупресивної терапії. Тяжкий перебіг хвороби та вимушена поліпрагмазія зумовили розвиток її численних ускладнень (сепсис, перфорація дванадцятипалої кишки, некротична пневмонія, гіпертрофія міокарда лівого шлуночка, дистрофічні зміни міокарда, печінки, нирок), несумісних із життям.
Результати аутопсії підтвердили клінічний діагноз. Зазначені зміни були потенційно очікуваними, оскільки, за даними попередніх дослідників, при застосуванні анти-В-лімфоцитарної терапії септицемії описані у 10–20% хворих [4, 9]. Отримувана хворим антибактеріальна терапія в таких ситуаціях недостатньо ефективна. Зазвичай наявність вторинного імунодефіциту у пацієнтів, які отримують ритуксимаб, рекомендується моніторувати шляхом контролю вмісту імуноглобулінів у сироватці крові [1], які в описаному випадку були достатніми і не потребували замісної терапії. Враховуючи дані аутопсії, більш показовою була оцінка субпопуляцій лімфоцитів периферичної крові, що може бути запропоновано для рутинного використання в подальшому.
Список використаної літератури
- 1. Barmettler S., Ong M.S., Farmer J.R. et al. (2018) Association of Immunoglobulin Levels, Infectious Risk, and Mortality With Rituximab and Hypogammaglobulinemia. JAMA Netw Open, Nov 2; 1(7): e184169. doi: 10.1001/jamanetworkopen.2018.4169. PMID: 30646343; PMCID: PMC6324375.
- 2. Bellutti Enders F., Bader-Meunier B., Baildam E. et al. (2017) Consensus-based recommendations for the management of juvenile dermatomyositis. Annals of the Rheumatic Diseases; 76: 329–340.
- 3. Kobayashi I., Akioka S., Kobayashi N. et al. (2020) Clinical practice guidance for juvenile dermatomyositis (JDM) 2018-Update, Modern Rheumatology, Volume 30, Issue 3: 411–423. doi.org/10.1080/14397595.2020.1718866.
- 4. Roberts J.E., Faino A.V., Bryan M.A. et al. (2024) Hospitalised infections and rituximab administration among children and adolescents with systemic lupus erythematosus from 2009 to 2021: Lupus Science & Medicine; 11: e001210.
- 5. Juvenile dermatomyositis (2021) Orphanet. www.orpha.net/en/disease/detail/93672.
- 6. Kim H. (2025) Juvenile Dermatomyositis: Updates in Pathogenesis and Biomarkers, Current Treatment, and Emerging Targeted Therapies. Pediatr Drugs 27, 57–72. doi.org/10.1007/s40272-024-00658-2.
- 7. Leclair V., Lundberg I.E. (2018) New Myositis Classification Criteria—What We Have Learned Since Bohan and Peter. Curr. Rheumatol. Rep., 20: 18. doi.org/10.1007/s11926-018-0726-4.
- 8. Solignac M., Cabrera N., Fouillet-Desjonqueres M. et al. (2024) JAK inhibitors in refractory juvenile rheumatic diseases: Efficacy, tolerance and type-I interferon profiling, a single center retrospective study, Journal of Autoimmunity, Volume 147:103248. ISSN 0896-8411, doi.org/10.1016/j.jaut.2024.103248.
- 9. McAtee C.L., Lubega J., Underbrink K. et al. (2021) Association of Rituximab Use With Adverse Events in Children, Adolescents, and Young Adults. JAMA Netw Open; 4(2): e2036321. doi:10.1001/jamanetworkopen.2020.36321.
- 10. Mondal S., Barman P., Vignesh P. (2022) Cardiovascular Abnormalities in Juvenile Dermatomyositis: A Scoping Review for the Clinical Rheumatologists. Front. Med. (Lausanne). Jun 24; 9: 827539. doi: 10.3389/fmed.2022.827539. PMID: 35814777; PMCID: PMC9263083.
- 11. Abu-Rumeileh S., Marrani E., Maniscalco V. et al. (2023) Lung involvement in juvenile idiopathic inflammatory myopathy: A systematic review/ Autoimmunity Reviews.Volume 22, Issue 10: 103416. ISSN 1568-9972. doi.org/10.1016/j.autrev.2023.103416.
- 12. Singh S., Suri D., Aulakh R. (2014) Mortality in children with juvenile dermatomyositis: two decades of experience from a single tertiary care centre in North India. Іnternational League of Associations for Rheumatology (ILAR). www.researchgate.net/publication/264164291_Mortality_in_children_with_juvenile_dermatomyositis_two_decades_of_experience_from_
a_single_tertiary_care_centre_in_North_India. - 13. Xiangyuan C., Xiaoling Z., Guangchao S. et al. (2024) Juvenile dermatomyositis complications: navigating gastrointestinal perforations and treatment challenges, a case report. Front Pediatr. Jul 12; 12: 1419355. doi: 10.3389/fped.2024.1419355. PMID: 39070551; PMCID: PMC11272553.
- 14. Xu Y., Ma X., Zhou Z. et al. (2021) Gastrointestinal perforation in anti-NXP2 antibody-associated juvenile dermatomyositis: case reports and a review of the literature. Pediatr. Rheumatol., 19: 2. doi.org/10.1186/s12969-020-00486-x.
Відомості про авторів
Ошлянська Олена Анатоліївна — докторка медичних наук, професорка, Національний університет охорони здоров’я України імені П.Л. Шупика, кафедра педіатрії, неонатології, дитячих інфекційних хвороб, імунології та алергології; ДУ «Всеукраїнський центр материнства та дитинства НАМН України».
ORCID ID: 0000-0002-9782-9709
Степанюк Ярина Степанівна — лікарка-педіатриня педіатричного відділення НДСЛ «Охматдит» МОЗ України.
ORCID ID: 0009-0003-3898-4703
Руденко Сергій Миколайович — кандидат медичних наук, завідувач педіатричного відділення НДСЛ «Охматдит» МОЗ України.
ORCID ID: 0000-0001-5935-3335
Лейбюк Юлія Василівна — лікарка-кардіоревматологиня нефрологічного відділення КНП «Івано-Франківська обласна дитяча клінічна лікарня».
ORCID ID: 0009-00072789-4234
Шатрова Клавдія Михайлівна — кандидатка медичних наук, доцентка кафедри морфології, клінічної патології та судової медицини, Національний університет охорони здоров’я України імені П.Л. Шупика.
ORCID ID: 0000-0003-2916-5098
Савостікова Наталія Леонідівна — лікарка-патологоанатоминя, відділення дитячої патологічної анатомії з відділом імуногістохімічних досліджень НДСЛ «Охматдит» МОЗ України.
ORCID ID: 0000-0003-1300-0248
Слободян Сергій Володимирович — лікар-рентгенолог центру радіології НДСЛ «Охматдит» МОЗ України.
ORCID ID: 0009-0001-9637-5426
Синоверська Ольга Богданівна — докторка медичних наук, професорка, завідувачка кафедри дитячих хвороб післядипломної освіти Івано-Франківського національного медичного університету.
ORCID ID: 0000-0003-1072-3782
Надійшла до редакції/Received: 12.03.2025
Прийнято до друку/Accepted: 25.03.2025
Leave a comment