Сучасний погляд на проблему аортального стенозу
Коваленко В.М., Полєнова Н.С., Тітов Є.Ю., Даниленко О.О.
Резюме. У статті наведено огляд сучасної літератури, що стосується етіології та патогенезу аортального стенозу, сучасних методів діагностики (магнітно-резонансна томографія, комп’ютерна томографія серця та 3D-ехокардіограма) та можливості використання традиційних та інноваційних методів лікування.
У розвинених країнах аортальний стеноз (АС) — найпоширеніша клапанна вада серця, що виникає у 3% осіб похилого віку і призводить до вищої смертності, ніж інші вади серця [40]. За даними різних авторів АС виявляють у 3–7% пацієнтів у загальній популяції, поширеність його підвищується з віком і в осіб старше 80 років досягає 15–20% [4]. За останні 20 років зареєстровано значне збільшення частки пацієнтів літнього віку [29]. Паралельно поширюється і популяція хворих із набутими вадами серця. При цьому пацієнти літнього віку — це особлива група з високим рівнем коморбідності, що нерідко визначає несприятливий прогноз і труднощі у виборі тактики лікування [4].
У дослідженні Cardiovascular Health Study, яке включало 5201 особу (чоловіки та жінки), вік яких старше 65 років, виявлено аортальний склероз у 26% учасників, у 2% усіх пацієнтів — виражений АС [4]. Поширеність АС майже в 2 рази вища у чоловіків, ніж у жінок, а расова приналежність не впливає на його поширеність [10]. Іншими факторами ризику появи АС є підвищення ліпопротеїдів низької щільності (ЛПНЩ) і холестерину, цукровий діабет, куріння, артеріальна гіпертензія [67]. Дегенеративний АС підвищує ризик виникнення інфаркту міокарда, порушення мозкового кровообігу, серцевої недостатності та раптової смерті. Дослідження останніх років виявили зв’язок між дегенеративним АС та порушеннями мінерального обміну [3, 5, 15, 53, 57].
Історія
Про можливості розвитку кальцинованих клапанів серця заговорили ще в середині XIX ст. У 1847 р. N. Lloyd під час аутопсії померлого хворого у віці 52 років із тяжким АС виявив, що стулки аортального клапана (АК) були осифіковані та пропускали лише кінчик зонда [27, 41].
У ХIХ ст. кальцифікати частіше виявляли при аутопсії хворих із вродженим двостулковим АК. У 1844 р. J. Paget припустив, що такий аномальний клапан у силу патологічного розвитку не просто має незвичайний зовнішній вигляд і недосконалу структуру, але і схильний до захворювань [32, 55].
У своїх класичних роботах, присвячених вивченню інфекційного ендокардиту, W. Osler неодноразово виявляв покритий вегетаціями двостулковий АК, але жодного разу не знаходив його стенозу і явищ вираженого кальцинозу стулок [52]. У 1923 р. T. Levis та R.T. Grant описали випадок «кальцієвого склерозу» в одного з 11 хворих із двостулковим АК, проте і вони не виявляли АС при інфекційному ендокардиті [38].
Звапніння АК було описано Йоганном Менкебергом на початку XX ст. у двох літніх чоловіків зі стенозом отвору АК. В обох випадках спостерігалася ізольована масивна петрифікація АК при інтактному мітральному клапані [46].
Патогенез
Останнім часом намітився перегляд ставлення до АС, що виникає у осіб літнього віку [1]. За кордоном цю ваду ніколи не пов’язували з атеросклерозом, а з часів опису Йоганном Менкебергом (1904) трактували ваду як дегенеративну, що виникає в результаті «зношування» АК з подальшою дистрофічною петрифікацією [43]. Але і ця концепція застаріла [2]. Буквально за кілька років нові дані висвітлили патологічний процес в АК як активне запалення та визначили нозологічно окреслене захворювання. При цьому показана роль клітинної, зокрема лімфоцитарної, інфільтрації, генетичної схильності до розвитку патологічного фіброзування в клапані, специфічних ендотеліальних пошкоджень, активації механізмів ектопічної осифікації тощо [9, 21]. З’явилися дані про дисфункцію ендотелію при хронічному запаленні, порушення мінералізації, пов’язаних із продукцією остеопонтину, ремоделюванням позаклітинного матриксу і персистуванням мікробних агентів [7, 24, 50, 58, 59, 71, 74, 75].
В останні роки різні дослідження показали спільний механізм патогенезу між дегенеративним АС і атеросклерозом [54]. Це узгоджується з гістопатологічними доказами того, що при ураженні АК залучені активні клітинні процеси, а самі запальні інфільтрати містять макрофаги, Т-клітини і гладком’язові клітини [11].
Роль місцевого і системного регулювання метаболізму кальцію і генетичних мутацій також вивчається в розвитку АС [45, 51]. Так, наявна більш висока поширеність кальцинуючої хвороби АК у пацієнтів із термінальною стадією ниркової недостатності, при хворобі Педжета та при гіперпаратиреозі. Крім того, мутації певних генів пов’язані з двостулковим АК і значною кальцифікацією аорти.
Причини
На сьогодні двома основними причинами АС є кальцифікація нормального тристулкового (раніше названа як сенільна чи дегенеративна кальцифікація) і вродженого двостулкового АК, при цьому АС прогресує незалежно від типу кальцифікації [35].
До кальцифікації АК призводить гостре запалення, ліпідні відкладення і механічне навантаження [53]. Цей патологічний процес має ті ж фактори ризику, що й при атеросклерозі, а саме: куріння, гіперхолестеринемія, гіпертонія, цукровий діабет, підвищення рівня креатиніну і кальцію в сироватці крові [70].
Кальцинуючий АС переважно спричинений відкладенням кальцію в стулках клапанів і злиттям їх комісур. Розташування цих відкладень допомагає пояснити варіабельність потоку крові при кальцинуючому АС, коли серцевий викид зростає при застосуванні інотропних препаратів або судинорозширювальних засобів [56].
Близько 1–2% дітей народжуються із двостулковим АК, що іноді поєднується з коарктацією аорти. Більшість із цих хворих немовлят належать до чоловічої статі. Двостулковий клапан становить більшу частку загального числа випадків АС, ніж тристулковий АК [61]. Процеси, які призводять до стенозу двостулкового АК, імовірно, аналогічні вищезазначеним для тристулкового клапана. Однак стеноз отвору при двостулковому клапані розвивається на два десятиліття раніше, ніж подібні зміни у тристулковому клапані аорти. Раннє прогресування можливе внаслідок менш сприятливої гемодинаміки при двостулковому клапані [17].
Природжений АС
У більшості випадків тяжкий вроджений АС виявляється і лікується в ранньому дитинстві або підлітковому віці. Іноді ця патологія діагностується вперше у зрілому віці. Стенокардія та серцева недостатність рідко наявні при вроджених АС, і пацієнти із безсимптомним АС більш схильні до раптової смерті, що пов’язано з появою гіпертрофії лівого шлуночка (ЛШ). Відсутність серцевої недостатності частково пояснюється тим фактом, що при існуючому перевантаженні тиском продуктивний викид довгий час зберігається за рахунок концентричної гіпертрофії ЛШ [20].
Дуже рідко причиною АС у розвинених країнах є ревматична хвороба серця [62]. Незважаючи на те що ревматична хвороба серця вражає в основному мітральний клапан, у ⅓ пацієнтів вона може розглядатися як ізольоване ураження АК [54]. Слід зазначити, що ревматичну хворобу АК окремо без ураження мітрального клапана виявляють вкрай рідко.
Таким чином, для встановлення діагнозу «ревматичний АС» необхідні типові ехокардіографічні докази ревматичної деформації мітрального клапана, оскільки АС при ревматизмі має спайкове злиття стулок, на відміну від кальцифікуючого АС (рисунок).
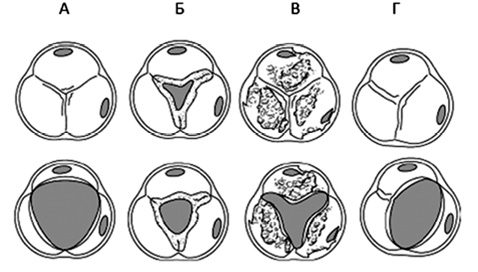
Рисунок. Морфологічні варіанти АС: А — інтактний АК; Б — АС ревматичної етіології; В — кальцифікація АК; Г — двостулковий АК
Патофізіологічні зміни при АС
Розвиток АС включає тривалий латентний період, протягом якого прогресивно погіршується функція ЛШ. Обструкція вихідного тракту призводить до гіпертрофії ЛШ [72]. Коли площа відкриття АК стає менше половини свого нормального розміру, підвищення градієнта тиску між ЛШ і висхідною аортою може бути виявлено за допомогою ехокардіографії чи шляхом прямої оцінки при катетеризації серця. Ці зміни відображають компенсаторне зростання тиску у ЛШ для підтримання адекватного системного тиску. Одним із наслідків цього є гіпертрофія ЛШ з подальшою діастолічною дисфункцією і стійко підвищеним тиском наповнення ЛШ [27]. Сильне скорочення лівого передсердя необхідне для забезпечення достатнього діастолічного наповнення ЛШ і підтримки адекватного ударного об’єму. На жаль, гіпертрофія має позитивні й негативні ознаки. Цей процес допомагає зберегти продуктивний викид, але погіршує коронарний кровотік, діастолічну функцію та підвищує смертність [23, 39].
При прогресуванні АС площа відкриття АК зменшується до ≤1 см² і структурно-функціональні зміни ЛШ більше не можуть компенсувати перешкоду відтоку і підтримувати систолічну функцію ЛШ на належному рівні. Гіпертрофія ЛШ при АС також веде до збільшення потреби міокарда в кисні [31]. Одночасно гіпертрофія міокарда ЛШ зумовлює стискання інтрамуральних коронарних артерій, які несуть кров до ендокарда. Ці зміни поряд зі зниженням діастолічного наповнення коронарних артерій зумовлюють появу класичної стенокардії, навіть у разі відсутності ішемічної хвороби серця (ІХС) [42]. Крім того, коли АС стає вираженим, серцевий викид не збільшується навіть при фізичному навантаженні. При цих умовах зниження периферичного судинного опору, який зазвичай відбувається при навантаженні, може призвести до артеріальної гіпотензії та непритомності [26, 34].
Зниження артеріального тиску під час фізичних вправ відзначено у пацієнтів з АС [64]. Деякі дослідники припустили, що дуже високий внутрішньошлуночковий тиск, який розвивається під час фізичних вправ у людей з АС, викликає рефлекторну депресорну відповідь, що, в свою чергу, призводить до непритомності. Зрештою, у деяких людей з АС провокувати непритомність при фізичному навантаження може шлуночкова тахікардія. Такі люди знаходяться у групі ризику післяопераційних рецидивів аритмії і мають бути розглянуті в подальшому кандидатами для електрофізіологічного тестування та можливої імплантації дефібрилятора [44].
Діагностика
Медичний огляд
АС можна запідозрити при об’єктивному обстеженні шляхом виявлення при аускультації типового систолічного шуму «crescendo-decrescendo», що іррадіює в шию. При легкій формі захворювання пік шуму припадає на початок систоли, II тон фізіологічно розщеплюється та на сонних артеріях не вислуховується. Коли потовщення клапана не викликає помітних перешкод для відтоку крові, говорять про аортальний склероз. Хоча сам по собі він доброякісний, наявність склерозу АК пов’язана з істотним підвищенням ризику серцевої смерті [53]. Оскільки АС та ІХС, швидше за все, мають схожі патофізіологічні механізми розвитку, аортальний склероз може бути раннім маркером ІХС і АС.
Функціональна діагностика
Проведення електрокардіограми (ЕКГ) у хворих з АС не має діагностичної цінності. Нормальна ЕКГ не виключає наявності тяжкого ураження клапана. У 85% пацієнтів із вираженим АС реєструють ЕКГ-ознаки гіпертрофіі ЛШ, однак між змінами на ЕКГ і ступенем тяжкості стенозу чіткого зв’язку немає. У пацієнтів без клінічних ознак АС проведення проби з фізичним навантаженням дозволяє уточнити функціональний стан серця і визначити прогноз перебігу захворювання так само, як і у пацієнтів з ІХС. За даними М.С. Amato та співавторів при негативному результаті проби 2-річна виживаність пацієнтів досягає 85%, а при позитивному результаті — лише 19% [8]. Результати декількох досліджень показали перевагу навантажувального тестування у осіб із безсимптомним тяжким АС. Р. Das та співавтори відзначили, що у >⅓ цих пацієнтів симптоми захворювання виникають під час фізичних вправ [19].
Рентгенограма грудної клітки при АС — неспецифічна. Серце зазвичай набуває типової форми у вигляді концентричної гіпертрофії ЛШ. Зрідка кальцифікацію АК можна побачити у бічних проекціях.
Трансторакальна ехокардіографія — один із найкорисніших методів для діагностики AС. Ехокардіографія з використанням режиму допплерографії служить основою для встановлення діагнозу АС. Це дослідження оцінює функцію ЛШ, ступінь гіпертрофії, кількість клапанних кальцифікатів, градієнт тиску і площу отвору відкриття АК, що необхідно для визначення прогнозу та лікування. Допплерівська оцінка швидкості потоку крові може бути також використана для моніторингу прогресування захворювання.
Більш чітка візуалізація отвору відкриття АК і планіметрична оцінка тяжкості АС можливі при проведенні черезстравохідної ехокардіографії [15]. Відповідно до рекомендацій АСС/АНА щодо ведення пацієнтів із клапанними вадами виділяють три ступені тяжкості АС (таблиця) [1].
| Ступіньтяжкості АС | Швидкість,м/с | Площа отвору,см | Середній градієнт тиску, мм | Індекс площі аортального отвору, см/м | Спостереження при трансторакальній ехоКГ |
|---|---|---|---|---|---|
| Норма | 1,5 | 3,0—4,0 | 5 | — | — |
| Легкий | <3,0 | >1,5 | <25 | — | 3—5 років за наявності зміни скарг або симптомів |
| Помірний | 3,0—4,0 | 1,0—1,5 | 25—50 | — | 1—2 роки за наявності зміни скарг або симптомів |
| Тяжкий | >4,0 | <1,0 | >50 | <0,6 | Щорічно за наявності зміни скарг або симптомів |
При ехокардіографічній оцінці тяжкості АС найчастіше використовують три параметри, а саме: розрахунок площі отвору відкриття клапана, визначення градієнта тиску і максимальної швидкості потоку. Регулярне спостереження пацієнтів із безсимптомним AС за допомогою ехокардіографії залежить від тяжкості стенозу. Ехокардіографічний моніторинг необхідно проводити щорічно у пацієнтів із тяжким AС, кожні 1–2 роки — при помірному AС та кожні 3–5 років — при незначному AС. Повторне ехокардіографічне обстеження слід також проводити при зміні клінічного статусу пацієнта [73].
Останнім часом ехокардіографічна оцінка тяжкості АС доповнена новими показниками: опір стулок АК, частка втрати ударної роботи ЛШ, коефіцієнт зниження кінетичної енергії потоку, стенотичне співвідношення, а також інтегральний індекс тяжкості АС, що становить суму ступенів кальцифікації АК (від 1 до 6) і рухливості стулок клапана (від 1 до 4) [12–65]. 3D-ехокардіограма (ехоКГ) дозволяє підвищити точність вимірювань АС та субаортального стенозу [32]. Успішна планіметрична оцінка площі АК при проведенні 3D-ехоКГ можлива у 92% осіб, і результати корелюють із даними, отриманими при виконанні черезстравохідної 2D-ехоКГ [69]. При використанні рівняння нерозривності з метою визначення площі АК беруть до уваги, що площа виносного тракту ЛШ має форму кола. Результати проведення 3D-ехоКГ показали, що на поперечних зрізах виносний тракт не завжди має форму кола, часто представлений у формі еліпса. Ця особливість лежить в основі недооцінки істинної величини площі відкриття АК. Інші автори замість використання рівняння нерозривності пропонують рутинно проводити пряме планіметричне вимірювання площі АК на основі 3D-ехоКГ. При цьому результати вимірювань були зіставними з даними інвазивного обстеження [69].
Ще одним варіантом підвищення точності визначення площі АК при використанні 3D-ехоКГ є отримання величини ударного об’єму в рівнянні нерозривності на основі визначення об’ємних параметрів ЛШ [33]. Ударний об’єм, отриманий у такий спосіб, є вищим порівняно з 2D-ехоКГ та схожий із даними магнітно-резонансної томографії (МРТ).
Стрес-ехокардіографія
Фармакологічний (добутаміновий) стрес-тест допомагає оцінити характер і ступінь тяжкості вади як у пацієнтів без клінічних ознак захворювання, так і за наявності систолічної дисфункції ЛШ, а також дає змогу прогнозувати перебіг захворювання, розвиток ускладнень і необхідність оперативного лікування.
Також проба з фізичним навантаженням чи фармакологічний тест дозволяють стратифікувати клінічні фактори ризику у пацієнтів із безсимптомним АС. Збільшення середнього градієнта тиску на АК на ≥18 мм рт. ст. є незалежним предиктором розвитку серцево-судинних ускладнень [36].
Але проведення навантажувальних тестів, які могли б уточнити тяжкість АС, протипоказане пацієнтам із наявними клінічними проявами.
Нові методи променевої діагностики
Впровадження у клінічну практику спіральної комп’ютерної томографії дозволяє проводити швидке й точне кількісне визначення кальцифікатів АК, аорти і коронарних артерій [6]. Індекс кальцію, що відображає щільність і розподіл депозитів кальцію в АК, може свідчити про тяжкість дегенеративного АС: чим він вищий, тим достовірно менша буде площа відкриття АК і вищий градієнт тиску.
Для уточнення функціонального стану АК застосовують ще один неінвазивний метод діагностики — МРТ.
Дослідження останніх 3 років показали, що значення площі відкриття АК, визначені при МРТ, черезстравохідній ехоКГ і катетеризації серця зіставні, коефіцієнти кореляції між ними сягають 0,83–0,96. Тому з 2005 р. МРТ рекомендовано для візуалізації структур АК, визначення площі отвору відкриття стулок АК планіметричним методом при неадекватній трансторакальній ехоКГ-візуалізації чи неможливості проведення черезстравохідної ехоКГ [16, 30].
Біомаркери
Мозковий натрійуретичний пептид (BNP) є маркером підвищення переднавантаження, тому рівень BNP корелює з вираженістю гіпертрофії ЛШ при АС [13]. Результати досліджень показали, що у симптоматичних пацієнтів рівень BNP вищий, ніж у пацієнтів без симптомів. Крім того, у осіб, в яких симптоми розвивалися незабаром після визначення рівня BNP, відзначено вищі концентрації пептиду порівняно з пацієнтами, в яких і в подальшому симптоми були відсутніми. Таким чином, BNP може стати корисним маркером для прогнозування появи симптомів, що, можливо, дасть змогу визначати необхідність проведення хірургічного втручання у безсимптомних пацієнтів. На жаль, наявність хронічного захворювання нирок, легеневої гіпертензії, ожиріння знижує прогностичну цінність BNP [28].
Катетеризація серця
Оскільки більшість пацієнтів з АС є особами старшого віку з підвищеним ризиком наявності ІХС, коронаровентрикулографія має бути проведена до хірургічного втручання, оскільки під час заміни АК може бути проведена реваскуляризація міокарда. У зв’язку з широкою доступністю неінвазивних методів діагностики в наш час проводити катетеризацію серця для оцінки гемодинаміки при АС не рекомендується [25].
Лікування
Тактика лікування пацієнтів з АС, на відміну від багатьох інших вад серця, зумовлена клінічною симптоматикою захворювання, і при появі симптомів необхідно вирішувати питання про хірургічну корекцію вади. Поява будь-якого нового симптому з боку серцево-судинної системи у пацієнта з діагностованим АС є причиною направлення його на консультацію до кардіохірурга. Більше того, є дані про переваги хірургічного втручання у пацієнтів із тяжким АС без клінічної маніфестації, особливо у осіб літнього віку [13].
У рекомендаціях з лікування пацієнтів із безсимптомним кальцинозом і стенозом отвору відкриття АК загалом відсутні показання до медикаментозної терапії. За наявності відповідних симптомів застосування різних препаратів може бути спрямоване на їх усунення, однак це не покращує прогнозу захворювання і нерідко обмежене гемодинамічними особливостями вади [22].
Медикаментозне лікування
Виходячи з сучасних уявлень про патофізіологічні механізми розвитку АС, а саме активний запальний процес, схожий на той, що відзначають при атеросклерозі, застосування інгібіторів 3-гідрокси-3-метилглутарил-КоА-редуктази — статинів — може мати певний позитивний вплив на перебіг захворювання [48–49]. Результати декількох ретроспективних досліджень показали, що у пацієнтів, які отримували статини, мало місце більш повільне прогресування стенозу, ніж у осіб, які їх не отримували [49, 60]. Проте у рандомізованому дослідженні, яке включало пацієнтів із помірним і тяжким АС, S.J. Cowell та співавтори не виявили позитивних ефектів від застосування статинів [18]. З іншого боку, L.M. Moura та співавтори при призначенні 20 мг розувастатину пацієнтам з АС та середньою величиною ЛПНЩ ≥8,8 ммоль/л спостерігали сповільнення темпів прогресування АС [47]. Додатковою перевагою статинів є здатність безпосередньо покращувати діастолічну функцію ЛШ [60].
За іншими даними у медикаментозному лікуванні АС статинами та інгібіторами ангіотензинперетворювального ферменту не виявили переваг у пацієнтів з АС [63].
Хірургічне лікування
Як європейські, так і американські експерти вважають, що єдиним методом лікування, здатним достовірно поліпшити прогноз і подовжити життя хворого із симптоматичним АС, є хірургічна корекція вади. Вибір тактики ведення пацієтів зводиться до застосування одного з трьох методів хірургічного лікування. Основними залишаються протезування АК (ПАК) — метод із найкращими показниками віддаленої виживаності — й балонна вальвулопластика. Останній має ряд недоліків, зокрема високий ризик ускладнень і значну ймовірність рецидиву обструкції, високу інтраопераційну летальність (>6%) і смертність протягом 1 року (25%) [1]. Однак у пацієнтів літнього віку внаслідок ряду причин цей паліативний вид лікування застосовують досить часто.
На сучасному етапі хірургічного лікування пацієнтів з АС з’явився новий перспективний метод — транскатетерна імплантація АК (ТІАК), який відкриває нові можливості для лікування пацієнтів із тяжким симптоматичним АС, яких раніше визнавали неоперабельними у зв’язку з тяжким клінічним станом і високим рівнем коморбідності. Ця нова технологія привертає величезну увагу кардіохірургів у всьому світі. У 2010 р. опубліковано результати рандомізованого проспективного дослідження PARTNER, яке стало першим із великих багатоцентрових досліджень, спрямованих на вивчення можливостей застосування ТІАК у пацієнтів із тяжким АС, які з різних причин визнані невідповідними кандидатами для стандартного ПАК [37].
Результати дослідження PARTNER продемонстрували переваги ТІАК перед стандартною консервативною терапією у пацієнтів із тяжким АС, визнаних неоперабельними. Необхідне подальше спостереження хворих, оскільки дані про тривалість функціонування клапана, встановленого за допомогою ТІАК, дуже обмежені. В окремих клінічних дослідженнях тривалість спостереження становила 5–8 років, проте передбачається, що для отримання достатньої кількості об’єктивної інформації знадобиться ≥10 років [66]. Строки для оперативного втручання мають вирішальне значення для заміни АК. На сьогодні рекомендують заміну АК у симптомних пацієнтів із тяжким АС, осіб з помірним АС, в яких проводять інші операції на серці (аортокоронарне шунтування, пластика висхідної аорти, протезування інших клапанів) або безсимптомних хворих із тяжким АС зі зниженою фракцією викиду ЛШ [14]. Іншим показанням до проведення хірургічної корекції вади у безсимптомних пацієнтів є поява симптомів при виконанні тесту з фізичним навантаженням.
Безсимптомні пацієнти мають бути проінформовані про високу ймовірність потреби хірургічного втручання в межах від 5 до 10 років, що зумовлено прогресуванням АС від помірного до тяжкого, а також про всі переваги та недоліки кожної з тактики ведення пацієнта. Консультування має включати обговорення ризику оперативної смертності, яка становить 3,2% при ізольованому ПАК та 5,6% — при поєднанні ПАК з аортокоронарним шунтуванням [63].
Таким чином, попри певні розбіжності експертів щодо етіології та патогенезу АС неревматичного генезу, не викликає сумнівів необхідність своєчасного виявлення вади, оцінки стану гемодинаміки та структурних змін серця з використанням усього арсеналу неінвазивних сучасних методів діагностики, що дозволить провести стратифікацію ризику та вибрати оптимальний спосіб корекції вади.
список використаної Літератури
1. Егоров И.В. (2010) «Старческий» порок сердца: истина и мифы. Леч. врач, 10: 32–36.
2. Егоров И.В., Шостак Н.А. (2000) Сенильный аортальный стеноз: современный взгляд на старую проблему. Клин. геронтология, 6(11–12): 37–42.
3. Карпова Н.Ю., Шостак Н.А., Рашид М.А. и др. (2006) Состояние костного метаболизма у больных з кальцинированным аортальным стенозом дегенеративного генеза. Клиницист, 1: 18–22.
4. Коваленко В.Н., Несукай Е.Г., Титов Е.Ю. (2010) Приобретенный аортальный стеноз: вопросы этиологии и патогенеза (url: http://www.rql.com.ua/cardio_j/2010/1/kovalenko.html).
5. Назаренко Г.И., Андропова О.В., Анохин В.Н. (2006) Дегенеративный (кальцинированный) аортальный стеноз, атеросклероз и остеопороз: клинико-морфологические параллели. Клиницист, 1: 11–17.
6. Черняков Р.М, Полубенцева Е.И., Андропова О.В. и др. (2005) Многосрезовая компьютерная томография в диагностике кальциноза клапанов сердца. Медвизуализация, 4: 111–115.
7. Agmon Y., Khandheria B.J., Tajik J.A. et al. (2004)Inflammation, infection and aortic valve sclero-sis. Insight from the Olmsted County (Minnesota) Population. Atherosclerosis, 174: 337–342.
8. Amato M.C., Moffa P.J.,Werner K.E. et al. (2001) Treatmentdecision in asymptomatic aortic valvestenosis: Role of exercise testing. Heart, 86: 381–386.
9. Anderson J.L., Lutz J.R., Gilbert E.M. et al. (1993) A randomized trial of low-dose beta-blockage therapy for idiopathic dilated cardiomyopathy. Lancet, 342: 1441–1446.
10. Aronow W.S., Ahn C., Kronzon I. et al. (2001) Association of coronary risk factors an duse of statins with progression of mild valvular aortic stenosis in older persons. Am. J. Cardiol., 88: 693–695.
11. Baumgartner H., Hung J., Bermejo J. et al. (2009) American Society of Echocardiography; European Association of Echocardiography. Echocardiographic assessment of valve stenosis: EAE/ASE recommendations for clinical practice [published correction appears in J Am SocEchocardiogr. 22(5): 442]. J. Am. Soc. Echocardiogr., 22(1): 1–23.
12. Bednarz J.E., Krauss D., Lang R.M. (1996)An echocardiographic approach to theassessment of aortic stenosis. J. Am. Soc. Echocardiogr., 9: 286–294.
13. Bergler-Klein J., Klaar U., Heger M. et al. (2004) Natriuretic peptides predictsymptom-free survival and postoperative outcome in severe aorticstenosis. Circulation, 109:2303–2308.
14. Bonow R.O., Carabello B.A., Chatterjee K. et al. (2008) 2006 Writing Committee Members; American College of Cardiology/American Heart Association Task Force. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation, 118(15): e523–e661.
15. Braunwald E. (2005)Heart disease: a text book of cardiovascular medicine. 7 ed. Philadelphia, ElsevierSaunders.
16. Caruthers Sh.D., Lin Sh.J., Brown P. et al. (2003) Practical value of cardiac magneticresonance imaging for clinical aorticvalve stenosis. Circulation, 108: 2236–2243.
17. Choo S.J., McRae G., Olomon J.P. et al. (1999) Aortic root geometry: pattern of diferences between leaflets and sinuses of valsalva. J. Heart Valve Dis., 8:407–415.
18. Cowell S.J., Newby D.E., Prescott R.J. et al. (2005)A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N. Engl. J. Med., 352: 2389–2397.
19. Das P., Rimington H., Chambers J. (2005) Exercise testing to stratify risk in aortic stenosis. Eur. Heart J., 26: 1309–1313.
20. Donner R., Carabello B.A., Black I. et al. (1983) Left ventricular walln stress in compensated aortic stenosis in children. Am. J. Cardiol., 51:946–951.
21. Fantini E., Demaison L., Sentex E. et al. (1994) Some biochemical aspects of the prospective effect of trimetazidine on rat cardiomyocytes during hypoxia and reoxygenation. J. Mol. Cell Cardiol., 26: 949–958.
22. Gaasch W., Schick E. Natural history of aortic stenosis (http://www.uptodate.com/contents/natural-history-of-asymptomatic-aorticstenosis-in-adults).
23. Gaasch W.H., Zile M.R., Hoshino P.K. et al. (1990)Tolerance of the hypertrophic heart to ischemia: studiesin compensated and failing dog hearts with pressure overloadhypertrophy. Circulation, 81:1644–1653.
24. Glader C.A., Birgander L.S., Soderberg S. et al. (2003) Lipoprotein (a), Chlamidiapneumoniae, leptin and tissue plasminogen activator as risk markers for valvular aortic stenosis. Eur. Heart J., 24(2): 198–208.
25. Gorlin R., Gorlin S.G. (1951) Hydraulic formula for calculation of the area of the stenotic mitral valve, other cardiac valves, and central circulatory shunts. Am. Heart J., 41:1–29.
26. Grech E.D., Ramsdale D.R. (1991)Exertional syncope in aortic stenosis: evidenceto support inappropriate left ventricular baroreceptor response. Am. Heart J., 121(2 pt. 1): 603–606.
27. Hess O.M., Ritter M., Schneider J. et al. (1984) Diastolic stiffness and myocardial structure in aortic valve disease before and after valve replacement. Circulation, 69(5): 855–865.
28. Horwich T.B., Hamilton M.A., Fonarow G.C. (2006) B-type natriuretic peptide levels in obese patients with advanced heart failure. J. Am. Coll. Cardiol., 47: 85–90.
29. Ivanovic B., Tadic M., Dincic D. (2010) The effects of arterial hypertension on aortic valve stenosis. Vojnosanit Pregl., 67(7): 588–559.
30. John A.S., Dill T., Brandt R.R. et al. (2003) Magnetic resonance to assess the aorticvalve area in aortic stenosis. How does itcompare to current diagnostic standards. J. Am. Coll. Cardiol., 42: 519–526.
31. Johnson L.L., Sciacca R.R., Ellis K. et al. (1978) Reduced left ventricular myocardial blood flow per unit mass in aortic stenosis.Circulation, 57(3): 582–590.
32. Khaw A.V., Bardeleben R.S., Strasser C. et al. (2009) Direct measurement of left ventricular out flow tract bytrans thoracic real-time 3D-echocardiography increases accuracy in assessment of aortic valve stenosis. Int. J. Cardiol., 136: 64–71.
33. Kizer J.R., Geffer W.B., de Lemos A.S. et al. (2001) Electron beam computed tomography for the quantification of aortic valvular calcification. J. Heart Valve Dis., 10: 361–366.
34. Kulbertus H.E. (1988) Ventricular arrhythmias, syncope and sudden death in aortic stenosis. Eur. Heart J., 9(Suppl. E): 51–52.
35. Kurtz C.E., Otto C.M. (2010)Aortic stenosis: clinical aspects of diagnosis and management, with 10 illustrative case reports from a 25-year experience. Medicine (Baltimore), 89(6): 349–379.
36. Lancelotti P., Lebois F., Simon M. et al. (2005) Prognostic importance of quantitative exercise Doppler echocardiography in asymptomatic valvular aortic stenosis. Circulation, 112(Suppl. I): 377–382.
37. Leon M., Smith C., Mack M. et al. (2010) Transcatheter aortic-valve implantation 23. for aortic stenosis in patients who cannot undergo surgery. N. Engl. J. Med., 363: 1597–1607.
38. Levis T., Grant R.T. (1923) Heart, 10: 21—26.
39. Levy D., Garrison R.J., Savage D.D. et al. (1990)Prognostic implications of echocardiographically determined leftventricular mass in the Framingham heart study. N. Engl. J. Med., 322: 1561–1566.
40. Lindroos M., Kupari M., Tilvis R. et al. (1993) Prevalence of aortic valve abnormalities in the elderly: an echocardiographic study of a random population sample. J. Am. Coll. Cardiol., 21(5): 1220–1225.
41. Lloyd N. (1847)Trans. Pathol. Soc. Lond., 1: 40.
42. Marcus M.L., Doty D.B., Hiratzka L.F. et al. (1982) Decreased coronary reserve: a mechanism for angina pectoris in patients with aortic stenosis and normal coronary arteries. N. Engl. J. Med., 307(22): 1362–1366.
43. Maridonneau-Parini I., Harpey C. (1990) Trimetazidine protects the human red blood cell against oxygen free radical damage. Cardiovasc. Drugs Ther., 4: 818–819.
44. Martinez-Rubio A., Schwammenthal Y., Schwammenthal E. et al. (1997) Patients with valvular heart disease presenting with sustained ventricular tachyarrhythmias or syncope: results of programmed ventricular stimulation and long-term follow-up. Circulation, 96: 500–508.
45. McKellar S.H., Tester D.J., Yagubyan M. et al. (2007)Sundt TM 3. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg., 134(2): 290–296.
46. Moonckeberg J.G. (1904) Virch. Arch. Pathol. Anat. Physiol. Klin. Med., 176: 472—496.
47. Moura L.M., Ramos S.F., Zamorano J.L. et al. (2007) Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J. Am. Coll. Cardiol., 49:554–561.
48. Ngo M.V., Gottdiener J.S., Fletcher R.D. et al. (2001) Smoking and obesity are associated with the progression of aortic stenosis. Am. J. Geriatr. Cardiol., 10:86–90.
49. Novaro G.M., Tiong I.Y., Pearce G.L. et al. (2001) Effect of hydroxymethylglutaryl coenzyme a reductaseinhibitors on the progression of calcifi c aortic stenosis. Circulation., 104: 2205–2209.
50. O’Brien K.D., Kuusisto J., Reichenbach D.D. et al. (1995) Osteopontin is expressed in human aortic valvular lesions. Circulation., 92(8): 2029–2032.
51. Ortlepp J.R., Hoffmann R., Ohme F. et al. (2001) The vitamin D receptor genotype predisposes to the development of calcific aortic valve stenosis. Heart, 85(6): 635–638.
52. Osler W. (1886) Trans. Assoc. Amer. Physicians, 2: 185—191.
53. Otto C.M., Lind B.K., Kitzman D.W.et al. (1999) Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N. Engl. J. Med., 341: 142–147.
54. Otto C.M., Bonow R.O. (2008): Valvular heart disease. Braunwald’s Heart Disease. 8 ed. Saunders Elsevier, Philadelphia, PA, р. 1625–1692.
55. Paget J. (1844) Trans. Roy. Med. Chir. Soc., 27: 162—171.
56. Paulus W.J., Sys S.U., Heyndrickx G.R. et al. (1991) Orifice variability of the stenotic aortic valve: evaluation before and after balloon aortic valvuloplasty. J. Am. Coll. Cardiol., 17:1263–1269.
57. Petty G.W., Khandheria B.K., Wisnant J.P. et al. (2000) Predictors of cerebrovascular events and death among patients with valvular heart disease. Stroke, 31: 2628–2635.
58. Poggianti E., Venneri L., Chubuchny V. et al. (2003) Aortic valve sclerosis with systemic endothe-lial dysfunction. J. Am. Coll. Cardiol., 41: 136–141.
59. Rajamannan N.M., Gersh B., Bonow R.O. (2003) Calcific aortic stenosis: from bench to the bedside — emerging clinical and cellular concepts. Heart, 89(7): 801–805.
60. Rajamannan N.M., Otto C.M. (2004)Targeted therapy to preventprogression of calcific aortic stenosis. Circulation, 110:1180–1182.
61. Roberts W.C., Ko J.M. (2005) Frequency by decades of unicuspid, bicuspid,and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation, 111:920–925.
62. Roberts W.C. (1970) Anatomically isolated aortic valvular disease: the casenagainst its being of rheumatic etiology. Am. J. Med., 49:151–159.
63. Rosseb A.B., Pedersen T.R., Boman K. et al. (2008) SEAS Investigators. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N. Engl. J. Med., 359(13): 1343–1356.
64. Schwartz L.S., Goldfischer J., Sprague G.J. et al. (1969) Syncope and sudden death in aortic stenosis. Am. J. Cardiol., 23: 647–658.
65. Shivelly B.K., Charlton G.A.,Crawford M.H. et al. (1998) Flow dependence of valve area in aortic stenosis: relation to valve morphology. J. Am. Coll. Cardiol., 31: 654–660.
66. Sinning J., Ghanem A., Steinhuser H. et al. (2010) Renal function as predictor of mortality in patients after percutaneous transcatheter aortic valve implantation. JACC Cardiovasc Interv., 3(11): 1141–1149.
67. Stewart B.F., Siscovick D., Lind B.K. et al. (1997) Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J. Am. Coll. Cardiol., 29(3): 630–634.
68. Supino P.G., Borer J.S., Preibisz J. et al. (2006) The epidemiology of valvular heart disease: a growing public health problem. Heart Fail. Clin., 2: 379–393.
69. Suradi H., Byers S., Green-Hess D. et al. (2010) Feasibility of using realtime «live 3D» echocardiography to visualizet he stenotic aortic valve. Echocardiography, 27: 1011–1020.
70. Taylor H.A.Jr., Clark B.L., Garrison R.J. et al. (2005) Relation of aortic valve sclerosis to risk of coronary heart disease in African-Americans. Am. J. Cardiol., 95:401–404.
71. Taylor P.M., Allen S.P., Dreger S.A. et al. (2002) Human cardiac valve interstitial cells in collagen sponge: a biological three-dimensional matrix for tissue engineering. J. Heart Valve Dis., 11: 298–307.
72. Tobin J.R.Jr., Rahimtoola S.H., Blundell P.E. et al. (1967) Percentage of left ventricular stroke work loss.A simple hemodynamic concept for estimation of severity in valvular aortic stenosis. Circulation, 35(5): 868–879.
73. Vahanian A., Baumgartner H., Bax J. et al. (2007) Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology; ESC Committee for Practice Guidelines. Guidelines on the management of valvular heart disease. Eur. Heart J., 28(2): 230–268.
74. Vehmaan-Kreula P., Puolakkainen M., Sarvas M. et al. (2001) Chlamidia pneumoniae proteins in-duce secretion of the 92-kDa gelatinase by human monocyte-derived macrophages. Athero-scler. Thromb. Vasc. Biol., 21(1): 1–8.
75. Yacoub M.H., Cohn L.H. (2004)Novel Approaches to Cardiac Valve Repair. From Structure to Function: Part I. Circulation, 109: 942–950.
Современный взгляд на проблему аортального стеноза
Резюме. В статье приведен обзор современной литературы, касающейся этиологии и патогенеза аортального стеноза, современных методов диагностики (магнитно-резонансная томография, компьютерная томография сердца и 3D-эхокардиограмма) и возможности использования традиционных и инновационных методов лечения.
аортальный стеноз, патогенез, чреспищеводная эхокардиография, трехмерная эхокардиография, транскатетерная имплантация клапана.
Адреса для листування:
Коваленко Володимир Миколайович
03680, Київ, вул. Народного ополчення, 5
ДУ «Національний науковий центр
«Інститут кардіології ім. М.Д. Стражеска»
НАМН України»
Leave a comment