АТЕРОСКЛЕРОЗ ЯК ЛІПІДНА ДИСТРОФІЯ І ЗАПАЛЕННЯ
Казимирко В.К., Иваницкая Л.Н. , Кутовой В.В., Дубкова А.Г. , Силантьева Т.С.
Резюме. Резюме. Атеросклероз розглядається як ліпідна дистрофія (холестериноз) і проліферативне гранулематозне запалення в інтимі судин. Наведено характеристику холестерину як індуктора запалення, а також самого запального процесу. Відзначена непродуктивність концепції про роль окиснювального «модифікування» в індукції запалення, відсутність ефекту від антиоксидантів при профілактиці та лікуванні атеросклерозу. Обґрунтована доцільність розробки антицитокінової терапії атеросклерозу.
Цель настоящей работы — ответить на вопросы: 1) почему из всех липидных дистрофий именно холестериноз лежит в основе развития атеросклероза; 2) какая особенность обмена холестерина (ХС) приводит к инфильтрации тканей, его накоплению в них; 3) какими особенностями, приводящими к индукции хронического воспаления в стенке сосуда, отличается молекула ХС; 4) как охарактеризовать это воспаление, если ориентироваться на классификации воспаления; 5) является ли оно иммунным; какие цитокины играют главную роль в воспалении при атеросклерозе; 6) имеет ли отношение к атерогенезу так называемое перекисное окисление липидов, окислительная «модификация» липопротеинов низкой плотности (ЛПНП); 7) какова роль в атерогенезе ферментативной «модификации» ЛПНП; 8) какие средства позволяют снизить активность атеросклеротического пролиферативного воспаления.
1. Атеросклероз как генетически обусловленная дистрофия. Исходя из общебиологических позиций атеросклероз следует рассматривать как дистрофию. Дистрофия — это морфологическое выражение нарушений тканевого (клеточного) обмена, ведущее к структурным изменениям, и, таким образом, она представляет собой один из видов повреждения. К морфогенетическим механизмам дистрофии относят инфильтрацию, декомпозицию, извращенный синтез, трансформацию [12, 13, 17]. Инфильтрация представляет собой избыточное проникновение продуктов обмена из крови и лимфы в клетки или межуточное вещество с последующим их накоплением в связи с недостаточностью ферментативных систем, метаболизирующих эти продукты. Инфильтрация ХС (в составе ЛПНП) интимы аорты и крупных артерий, ряда тканей при атеросклерозе является классическим примером [6]. В соответствии с классификацией дистрофий [12, 16, 17], атеросклероз — это общая жировая (холестериновая) мезенхимальная дистрофия приобретенного или наследственного характера, возникающая на фоне отсутствия в организме ферментных систем, метаболизирующих ХС. Их отсутствие в организме обусловлено генетически.
2. Уникальные особенности молекулы ХС и его обмена. Молекула ХС отличается уникальным сочетанием ряда структурных особенностей, в том числе имеет жесткий углеродный скелет циклопентанопергидрофенантрена, который не разрушается ферментными системами организма. Метаболизм ХС в организме хорошо сбалансирован, механизмы поддержания его баланса в тканях достаточно изучены [7]. Любое нарушение функционального состояния организма сопровождается изменением уровня ХС в крови. В стационарном состоянии здорового человека суммарное количество ХС, синтезированного в тканях и поступившего с пищей, равно суммарному количеству экскретируемых ХС и желчных кислот (ЖК). Баланс ХС представляют как разность между прибылью и убылью его в организме:
(ХСэнд. + ХСэкз.) — (ХСэкскр. + ЖКэкскр.) = 0.
Возможные варианты баланса ХС включают нулевой, отрицательный, положительный [8]. Из вышепредставленной формулы следует, что положительный баланс ХС в силу того, что его молекула не разрушается, будет сопровождаться повышением вероятности заболевания атеросклерозом и желчнокаменной болезнью. ХС откладывается не только в стенках артерий, но также в клапанах, сухожильных нитях сердца, коре надпочечников, пирамидах почек, сухожилиях мышц, хрящах, коже, роговой оболочке глаза и других тканях, то есть положительный баланс ХС проявляется в виде холестериноза. Увеличение общего количества ХС в организме по сравнению с потребностью в нем наблюдается при наличии так называемых факторов риска. Имеет значение не только количественное накопление ЛПНП, содержащих наибольшее количество ХС из всех его транспортных форм, но и качественное их состояние custom essay writing services reviews — размеры, плотность. Таким образом, в атерогенезе ведущую роль играет нарушение именно баланса ХС. Превышение количества ХС, необходимого для организма (синтез мембран, стероидных гормонов, витамина D3, ЖК), неизбежно будет сопровождаться его усиленным поступлением в интиму артерий (ровно как и в другие ткани) и отложением там с развитием воспаления вокруг внеклеточных кристаллов эфиров и свободного ХС, поскольку они являются инородными телами для этой ткани. Затруднительно говорить о нормальном уровне ХС в организме вообще, ибо любое превышение необходимого для организма его количества будет всегда иметь патологические последствия в виде атеросклероза: не подвергающийся утилизации в организме ХС будет накапливаться в интиме сосудов.
ХС поступает в организм из двух источников — с пищей и за счет эндогенного синтеза. ХС в тканях может синтезироваться из любых веществ, при распаде которых образуется ацетил-коэнзим А (ацетил-КоА). Генетические, гормональные и пищевые факторы могут изменять многие функции печени и влиять на баланс ХС в организме. Основными местами синтеза ЛП являются печень и слизистая оболочка тонкой кишки. Рецепторы ЛПНП, содержащиеся в печени, удаляют из крови 75% частиц ЛПНП. Уменьшение количества рецепторов сопровождается повышением уровня ЛПНП в плазме крови и ранним развитием атеросклероза, ишемической болезни сердца. Дефицит рецепторов может быть генетически обусловленным (семейная гиперхолестеринемия — ГХС) и приобретенным. Для максимальной экспрессии рецепторов ЛПНП на клеточных мембранах (что обеспечивает удаление частиц ЛПНП из крови) необходимы эстрогены, инсулин, тироксин. Избыточное потребление насыщенных жиров, ХС приводит к уменьшению продукции рецепторов. Главными носителями ХС являются ЛПНП и липопротеины очень низкой плотности (ЛПОНП), в ядре которых ХС присутствует в виде эфиров. Клетки имеют специфические для ЛПНП рецепторы [20]. Нарушение элиминации ЛПНП в связи с уменьшением числа или аномальным функционированием рецепторов ЛПНП, молекулярными дефектами рецепторного белка ведет к ГХС и атеросклерозу. От активности рецепторов зависит степень ГХС. Второй генетической причиной ГХС являются структурные изменения аполипопротеина В (апо-В), вследствие чего нарушается связывание с нормальными рецепторами.
Суточный оборот ХС составляет около 1 г. Он находится в состоянии динамического равновесия: количество синтезируемого и принятого с пищей соответствует выводимому количеству. Суточная потребность в ХС составляет около 1 г и может покрываться за счет биосинтеза. При смешанной диете почти суточная норма ХС синтезируется в кишечнике, коже, печени. Ежесуточно из организма выводится около 1 г ХС. Очень незначительная часть ХС используется для биосинтеза стероидных гормонов. ХС наряду с глюкозой, мочевой кислотой, щелочной фосфатазой относится к веществам, концентрация которых прогрессивно повышается на протяжении взрослой жизни. Уровень ХС в крови может и не отражать его содержание в организме, несмотря на то, что весь ХС тканей организма обладает способностью обмениваться с ХС в плазме крови [4]. В норме около 70% ХС в плазме крови находится в составе «атерогенных» ЛПНП и ЛПОНП, а в составе «антиатерогенных» липопротеинов высокой плотности (ЛПВП) циркулирует 30%. Баланс скоростей притока и оттока ХС в сосудистой стенке (и других тканях) сохраняется при таком соотношении. Выделены три пула ХС: быстро обменивающийся, промежуточный, медленно обменивающийся [18]. ХС каждого из пулов обменивается с ХС в плазме крови, при этом скорости установления равновесия сильно различаются.
В регуляции синтеза ХС в организме участвуют различные механизмы [7]: синтез ХС ингибируется ЛПНП при их связывании с соответствующими рецепторами апо-В-100. Гидрокси-метилглутарил коэнзим-А-редуктаза (ГМГ-Ко-А-редуктаза) ингибируется ХС по принципу обратной связи, ее активность повышает инсулин и тиреоидный гормон. Глюкагон и глюкокортикоиды уменьшают активность ГМГ-Ко-А-редуктазы, она снижается в период голодания. При увеличении поступления ХС с пищей уменьшается эндогенный синтез, но полного его прекращения не наблюдается (ингибируется только образование ХС в печени). Уменьшая количество ХС в пище, можно снизить его уровень в крови. Синтез ХС ингибируют богатые им остатки хиломикронов.
Печень — единственный орган, осуществляющий выведение ХС из организма (выделение его сальными железами и стенкой кишечника незначительно). В процессе катаболизма он превращается в ЖК и в составе желчи поступает в кишечник. ЖК обеспечивают растворимость ХС в желчи и способствуют перевариванию липидов. Кишечная микрофлора продуцирует ферменты, осуществляющие химическую модификацию ЖК. Из организма млекопитающих ХС выводится с калом главным образом в виде ХС, продуктов жизнедеятельности кишечной микрофлоры, копростанола, холестанола. Однако бо`льшая часть ЖК всасывается кишечным эпителием и после попадания в печень вновь секретируется в составе желчи (энтерогепатическая циркуляция ЖК). Поэтому из 15–30 г солей ЖК, ежедневно поступающих в кишечник с желчью, в экскрементах выявляют только около 0,5 г [5]. Это соответствует ежесуточному биосинтезу ХС de novo. Синтез ЖК из ХС регулируется по механизму отрицательной обратной связи, но молекулярный и биохимический характер этого процесса до конца не установлен. Критическим этапом в синтезе ЖК является микросомальное 7α-гидроксилирование ХС.
3. Атеросклероз как воспаление. Патофизиологически атеросклероз представляет собой пролиферативное гранулематозное воспаление, локализующееся в соединительной ткани (СТ) интимы сосудов и развивающееся в ответ на накопление в ней внеклеточно эфиров и свободного ХС в виде кристаллов. Биологический смысл воспаления заключается в ликвидации или отграничении очага повреждения и вызвавших его патогенных факторов [10], к которым относится и подлежащий выведению из организма такой продукт обмена веществ, как ХС. Выделяют альтеративное, экссудативное и пролиферативное (или продуктивное) воспаление. Из трех видов продуктивного воспаления (интерстициальное, гранулематозное, воспалительные гиперпластические (гиперрегенераторные) разрастания) [9] гранулематозное хроническое воспаление вызывается нерастворимыми или медленно разрушающимися раздражителями и сопровождается развитием гранулем. Гранулемы представляют собой компактное скопление макрофагов и/или эпителиоидных клеток. В число эндогенных этиологических факторов гранулем входят и продукты нарушенного обмена [14]. Различают иммунное и неиммунное пролиферативное гранулематозное воспаление [15]. Иммунное сопровождается образованием клеточно-опосредованных иммунных гранулем и антительно-опосредованных. Неиммунное гранулематозное воспаление включает большинство гранулем, образующихся вокруг инородных тел. Морфологически различают два основных вида гранулем: зрелые макрофагальные и эпителиоидно-клеточные [11]. Гранулемы, вызванные эндогенными жирами, относятся к гранулемам инородных тел [9]. В соответствии с определением W.L. Epstein [21], гранулема инородных тел является неиммунной реакцией мононуклеарных фагоцитов на нерастворимый эндогенный или экзогенный стимул. В неиммунных гранулемах, образующихся вокруг инородных тел, преобладают механизмы ауторегуляции за счет биологически активных веществ, синтезируемых макрофагами, — различных цитокинов, факторов роста, выделяемых макрофагами, гладкомышечными клетками (ГМК), лимфоцитами. Гранулематоз в целом отражает несостоятельность системы моноцитарных фагоцитов: фагоцитоз подменяется отграничением раздражителя (возбудителя) [12, 16]. Развитие гранулемы проходит в три стадии: 1) накопление мононуклеарных фагоцитов; 2) созревание их в макрофаги, агрегация; 3) созревание мононуклеарных фагоцитов в эпителиоидные клетки, слияние их или макрофагов в гигантские клетки [14]. Гистологически гранулемы могут быть чисто макрофагальными, то есть без многоядерных и эпителиоидных клеток [16], что имеет место при атеросклерозе. Для развития гранулемы необходимы три условия: 1) наличие веществ, способных стимулировать трансформацию моноцитов в макрофаги; 2) стойкость раздражителя; 3) высокая местная концентрация раздражителя [14, 19]. Главным условием развития гранулематоза является стойкость раздражителя по отношению к фагоцитам: гранулема существует, пока в ткани имеется раздражитель. Для этого раздражитель должен находиться в тканях в виде частиц, которые не подвергаются полной деградации макрофагами, привлекаемыми в очаг воспаления.
При атеросклерозе имеет место повреждающее действие внеклеточного ХС на клетки и межуточное вещество рыхлой СТ в субэндотелиальном пространстве сосудов. По данным экспериментальных исследований [1], стадия экссудации при инкорпорации ХС в интиму хорошо выражена в виде мукоидного отека. Активируются лизосомальные ферменты клеток СТ, происходит эластолиз, освобождение и поступление в межуточное вещество гликозаминогликанов. Главным условием развития атеросклеротического пролиферативного гранулематозного воспаления является стойкость ХС, становящегося эндогенным инородным телом для СТ интимы артерии. При развивающемся воспалении дирижером совокупности клеточных медиаторов являются цитокины макрофагов. Эти клетки являются посредниками привлечения клеток в воспалительную реакцию. Атеросклеротические гранулемы-бляшки содержат большое количество макрофагов, Т-клеток, ГМК, которые продуцируют коллаген, ограничивающий атероматозное ядро от «нормальной» ткани, в них наблюдается развитие сосудистых элементов (ангиогенез). При атеросклеротическом воспалении полного разрешения процесса нет, поскольку раздражитель — кристаллы ХС не подвергаются деструкции (в организме отсутствуют ферменты, которые бы расщепляли циклопентанопергидрофенантреновое ядро молекулы ХС), более того, продолжается дальнейшее поступление и накопление ХС ЛПНП и ЛПОНП в атероматозных бляшках. Эффективного удаления ХС частицами ЛПВП из интимы по лимфатическим путям не наблюдается. Нагруженные липидами макрофаги остаются в атеромах, они разрушаются и липиды (эфиры и свободный ХС в виде кристаллов) накапливаются внеклеточно, что в еще большей степени затрудняет их удаление из очага. Атеросклеротическое воспаление как ответ на инкорпорацию в интиму кристаллов ХС приводит к повреждению сосудистой стенки. При активации макрофагов, Т-лимфоцитов и разрушении протеолитическими ферментами «покрышки» атеросклеротической гранулемы воспаление переходит во вторую фазу: повреждается эндотелий, активируется свертывание крови, в сосудах образуются пристеночные или окклюзирующие тромбы, в крови появляются маркеры воспаления [22, 24], что используется с диагностической и прогностической целью. До этого момента поступления в систему кровообращения медиаторов межклеточных взаимодействий, как правило, не наблюдается.
Таким образом, патогенетически атеросклеротические «бляшки» представляют собой неиммунные гранулемы, образующиеся вокруг эндогенных «инородных» тел — жидких и твердых кристаллов ХС в СТ интимы артерий. Гистологически — это зрелые макрофагальные гранулемы различного размера без гигантских и эпителиоидных клеток, с вне- и внутриклеточно (в макрофагах) расположенными кристаллами ХС, фиброзными изменениями и нередким некрозом в центре. Воспаление лежит как в основе происхождения атеросклеротической бляшки, так и ее прогрессирования, эволюции в поврежденную бляшку, возникновения разрыва капсулы. При атеросклеротическом воспалении наблюдается характерная для воспаления межклеточная кооперация с продукцией цитокинов, пролиферация ГМК, образование молодой СТ в гранулемах, формирование ограничивающей (изолирующей) липидное ядро соединительнотканной капсулы. (Вокруг холестеринового ядра образуется защитный соединительнотканный барьер). В соответствии с рабочей классификацией гранулем А.И. Струкова и О.Я. Кауфман (1989) можно заключить, что по своей сущности атеросклеротический процесс представляет собой хроническое пролиферативное гранулематозное воспаление, вызванное эндогенным фактором — ХС. Морфологически атеросклеротические бляшки (атеромы) относятся к макрофагальным гранулемам. Патогенетически это неиммунные гранулемы, образующиеся вокруг эндогенных инородных тел — кристаллов ХС в интиме артерий. Гистологически атеросклеротические бляшки — это зрелые макрофагальные гранулемы различного размера без гигантских и эпителиоидных клеток, с внеклеточно расположенными эндогенными инородными частицами — кристаллами ХС, с фиброзными изменениями и нередким некрозом в центре [16].
Кристаллы ХС вызывают развитие воспаления не только в сосудистой стенке. Оно возникает как реакция на холестериновые камни в желчном пузыре или кристаллы ХС в тканях. При эссенциальной ГХС липиды, кристаллы ХС откладываются в различных тканях, в том числе в сухожильных влагалищах, суставных капсулах, что сопровождается возникновением реактивного воспаления, тендинита, синовита, артрита [3]. Этот процесс сопровождается появлением в крови маркеров воспаления. Возникающие в сухожилиях и периосте ксантомы, могут кальцифицироваться, обусловливать эрозии костей. В синовиальной жидкости, кроме большого количества полинуклеаров, выявляют «пенистые» клетки — макрофаги, вакуоли которых содержат липиды, в частности ХС или его эфиры. (Такие клетки имеются в норме и при патологии). Как мы отмечали, в очаге атеросклеротического воспаления они подвергаются апоптозу, в результате чего в интиме накапливается внеклеточный ХС, его кристаллы — индукторы атеросклеротического воспаления. Близким примером гранулематозного воспаления в организме человека, подобного атеросклеротическому, является подагра. Тофусы при подагре представляют собой воспалительные гранулемы, изолирующие эндогенные инородные тела — кристаллы уратов. Подобно ситуации с атеросклерозом, развивающимся вследствие отсутствия в организме ферментов, разрушающих циклопентанопергидрофенантреновое ядро ХС, в организме человека отсутствует фермент уриказа, превращающий мочевую кислоту в растворимое соединение. В результате происходит накопление мочевой кислоты, отложение кристаллов в тканях с развитием хронического пролиферативного гранулематозного воспаления. Таким образом, тофусы при подагре и «бляшки» при атеросклерозе — это гранулемы вокруг эндогенных инородных тел. Отложение кристаллов мочевой кислоты в тканях определяет клинические проявления. Кристаллы мочевой кислоты фагоцитируются клетками синовиальной оболочки, их активация сопровождается секрецией интерлейкина (ИЛ)-1, ИЛ-6, ИЛ-8, фактора некроза опухоли (ФНО)-α, притоком фагоцитов. Воспаление сопровождается формированием фиброза синовии, эрозией суставного хряща, анкилозами. Тофусы возникают в суставах, костях, под кожей и даже в сердце, гранулематозные поражения могут возникать в почках. При длительном снижении уровня мочевой кислоты в крови тофусы-гранулемы могут рассасываться.
Мы уже отметили, что кристаллы ХС вызывают развитие воспаления не только в сосудистой стенке. Отложение ХС в различных тканях при эссенциальной ГХС сопровождается ксантоматозом, резкой болью в животе, липемическим ретинитом, гепатоспленомегалией [3]. При гиперлипопротеинемии (ГЛП) IIа, реже — IV типа в результате отложения липидов в суставных и периартикулярных структурах развиваются артро- и тендопатии. При ГЛП IIа ксантомы возникают в сухожилиях и периосте, они могут кальцифицироваться, обусловливать эрозии костей. Возникает транзиторный или мигрирующий полиартрит, похожий на подагрический. В крови повышено содержание общего ХС, ХС ЛПНП, появляется С-реактивный белок, повышается скорость оседания эритроцитов. Тот факт, что при ГЛП IV типа реже, чем при IIа, отмечаются подобные проявления, подчеркивает роль ХС в генезе проявлений. При ГХС (особенно гетерозиготной форме семейной ГХС) отмечается липоидная дуга роговицы, ксантелазмы, ксантомы сухожилий, туберозные ксантомы на голенях и локтях, боль в суставах. Как и атеросклеротические гранулемы, ксантомы состоят из макрофагов, пенистых клеток, переполненных эфирами ХС. Вокруг возникает воспаление с присутствием гигантских клеток и синтезом коллагена. Таким образом, ксантомы, как и атеросклеротические бляшки, являются воспалительными гранулемами. Все ксантомы имеют потенциальную обратимость в случае снижения концентрации липидов в плазме крови. Так, при ГЛП I и V типа ксантомные высыпания могут в течение нескольких месяцев полностью исчезнуть. Ксантелазмы и бугорчатые ксантомы при выраженном снижении уровня липидов в плазме крови исчезают в течение нескольких лет. Наиболее трудно обратному развитию подвергаются сухожильные ксантомы.
О том, какие негативные последствия для тканей имеет накопление в них ХС, его нерасщепленных эфиров, свидетельствует такая патология, как болезнь Вольмана. При болезни Вольмана имеется генетический дефект — отсутствие в лизосомах кислой липазы, в результате накапливаются эфиры ХС в лизосомах печени, селезенки, надпочечников, гемопоэтической системы, тонких кишок с летальным исходом. Между этим заболеванием и атеросклерозом имеется определенная аналогия: в организме человека в норме отсутствует ферментная система, разрушающая молекулу ХС, здесь же имеется врожденный дефицит фермента кислой липазы.
4. Роль так называемого перекисного окисления липидов в атерогенезе. Возможность окисления ЛПНП in vivo в русле крови не доказана и маловероятна [18]. Поступающие в гемодинамически уязвимых местах в интиму ЛПНП подвергаются воздействию тканевых ферментов (ферментативному «модифицированию») и в дальнейшем поглощаются макрофагами. Окислительному «модифицированию» (воздействию активных форм кислорода) частицы ЛПНП могут подвергаться только внутри макрофагов. После их разрушения продукты окисления появляются в атеромах, где их и находят исследователи. Инициатором пролиферативного воспаления в интиме сосудов в действительности является неразрушающаяся циклопентанопергидрофенантреновая структура ХС, его кристаллы, а не окисленные ЛПНП. Концепция окислительного модифицирования не получила реального подтверждения практикой: антиоксиданты не замедляют прогрессирование атеросклероза. Еще в 1997 г. результаты 26 длительных широкомасштабных исследований, проведенных в соответствии с требованиями надлежащей клинической практики (GCP), показали негативные последствия профилактики сердечно-сосудистых заболеваний с помощью антиоксидантов. Последующие доказательные исследования только подтвердили этот факт. Так, отрицательные результаты лечебного и профилактического действия антиоксидантов при атеросклерозе получены в таких крупных исследованиях, как HPS, HOPE, HATS (ангиографическое исследование) [2]. При этом применение антиоксидантов больными уменьшало положительное влияние симвастатина и ниацина на ангиографический и клинический эффект.
5. Роль в атерогенезе макрофагов. Ведущую роль в атеросклеротическом процессе играют макрофаги, основной функцией которых является удаление различных веществ, клеток, их фрагментов и т.д. Фагоцитарная несостоятельность макрофагов обусловлена отсутствием лизосомальных ферментов, которые бы разрушали углеродное ядро ХС (в них лизируются только эфиры ХС). И, таким образом, главным условием развития атеросклеротического пролиферативного гранулематозного воспаления является стойкость внеклеточного ХС, становящегося эндогенным инородным телом для СТ интимы артерии. В атеромах-гранулемах повреждение имеет перманентный характер, деструктивные и восстановительные процессы протекают одновременно. Они содержат макрофаги/пенистые клетки, Т-лимфоциты, ГМК, которые продуцируют коллаген, ограничивающий атероматозное ядро от «нормальной» ткани. В сформированных гранулемах наблюдается развитие сосудистых элементов (ангиогенез). Клеточный компонент гранулем, расположенный в соединительнотканной капсуле, включает ГМК, макрофаги и лейкоциты. В зонах под капсулой и по бокам ее (в области «плеч») имеется смесь макрофагов, ГМК, Т-лимфоцитов. Пенистые клетки происходят преимущественно из моноцитов крови, превращающихся в стенке сосудов в макрофаги. Волокнистый компонент гранулем состоит из внеклеточного матрикса СТ, включающего в себе коллагеновые и эластические волокна, а также протеогликаны. Основная масса гранулемы — некротизированный центр (сердцевина), состоящий из кристаллов ХС и его эфиров, пенистых клеток, детрита (остатков клеток), белков плазменного происхождения и извести. Гранулемы-бляшки отличаются друг от друга по количеству составных частей каждого из трех структурных компонентов [12]. Развитая гранулема (атерома) содержит много липидов, иногда старая фиброзная гранулема построена преимущественно из ГМК и фиброзной ткани. При прогрессирующем фиброзе гранулемы превращаются в рубцы, деформирующие стенку артерии. В длительно существующих гранулемах в большом количестве может содержаться базофильная известь каменистой плотности, в основном содержащая фосфат кальция.
6. Последовательность морфогенеза атеросклеротических гранулем. Последовательность морфогенеза атеросклеротических гранулем состоит из накопления в СТ интимы сосуда ЛПНП, их повреждения ферментами («модификации»), проникновения (через посредство моноцитарного хемотаксического фактора и адгезивных молекул) моноцитов, трансформации этих клеток в макрофаги, а затем, по мере накопления липидов, — в пенистые клетки, миграции под действием цитокинов макрофагов (ФНО-α, ИЛ-1, тромбоцитарного фактора роста (PDGF)) в очаг воспаления ГМК из медиа, их пролиферации и продукции СТ, изолирующей внеклеточный ХС, формирования макрофагальных гранулем (простых гранулем, или фагоцитом), изолирующих ХС в интиме сосудов. Периоды и стадии атерогенеза представлены в таблице.
Таблица. Периоды и стадии атерогенеза
| Стадия | Основные морфологические изменения |
| Довоспалительный период | |
|---|---|
| I | Жировые пятна |
| II | Жировые полоски |
| Период хронического продуктивного (пролиферативного) гранулематозного воспаления |
|
| III | Переходные поражения (появление внеклеточного ХС в интиме) |
| IV | Начало пролиферативного гранулематозного воспаления и формирования гранулем |
| V | Сформировавшиеся гранулемы |
| VI | Исходы гранулем |
Атеросклероз как хроническое пролиферативное гранулематозное воспаление начинается с момента появления внеклеточных липидных депозитов, содержащих ХС. Небольшое количество внеклеточных липидов появляется на стадии так называемых переходных поражений и имеет ключевое значение для атерогенеза. Большое число экспериментальных исследований свидетельствует о поэтапной эволюции жировых полосок в развитые атероматозные гранулемы. Ключевое событие атерогенеза приведено на рисунке.
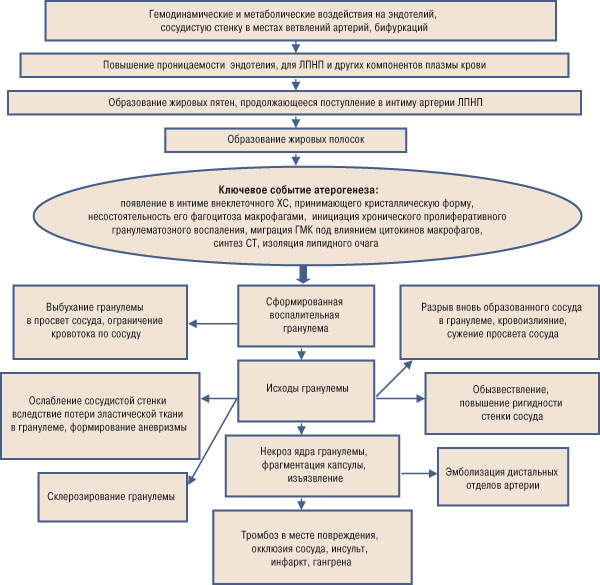
Рисунок. Ключевое событие атерогенеза
Таким образом, атеросклеротическое пролиферативное воспаление является ответом на появление внеклеточного ХС, его кристаллов в интиме артерий. В целом пролиферативное гранулематозное воспаление при атеросклерозе развивается в рамках деятельности системы моноцитарных фагоцитов. Оно является результатом воздействия внеклеточного ХС, его кристаллов, активирующих систему моноцитарных фагоцитов, или гистио-макрофагальную систему. Вокруг этого чужеродного агента в интиме скапливаются клетки макрофагального происхождения, образующаяся гранулема ограничивает раздражителя. Гранулема — одна из форм клеточной реакции, предназначенная для избавления организма от чужеродных начал. Таким чужеродным началом становится внеклеточный ХС, не подвергающийся (в отличие от других липидов) разрушению из-за отсутствия в организме необходимых ферментов. Незавершенность фагоцитоза тормозит обратное развитие гранулем-бляшек. Более того, поступление ХС в бляшки продолжается. После повреждения защитной оболочки — воспалительного барьера — гранулема становится источником цитокинов, выявляемых в периферической крови.
Как показывают приведенные выше сведения, атеросклероз как заболевание включает два важных проявления: 1) общую липидную дистрофию (холестериноз) и 2) следствие этой липидной дистрофии — локальный воспалительный процесс во внутренней оболочке артерий, а также в некоторых других тканях. Причиной холестериноза является положительный обменный баланс ХС — вещества, которое в организме не расщепляется, не растворяется, а обмен построен на балансе между синтезом и поступлением с одной стороны и выведением из организма (в основном в виде ЖК) — с другой. Это свидетельствует о генетической обусловленности данного заболевания. Причинами положительного баланса, а значит, прогрессирования атеросклероза, является целый ряд факторов приобретенного или наследственного характера, которые сегодня именуются факторами риска. Из веществ, которые из плазмы крови попадают в субэндотелиальное cell phone spy software пространство и депонируются там, триггером воспаления может быть только ХС, его кристаллы, поскольку все остальные вещества, попадающие из плазмы крови в СТ субэндотелия, в том числе другие липиды, в отличие от ХС, распадаются — метаболизируются. Только ХС, его циклопентанопергидрофенантреновая структура, не поддается разрушению. Концепция окислительного модифицирования ЛПНП не подтверждается практикой: антиоксидантная терапия при атеросклерозе неэффективна.
В настоящее время основным противовоспалительным средством при атеросклерозе являются статины. Антицитокиновая терапия атеросклероза (учитывая, что главным цитокином гранулематозного процесса является ФНО-альфа, а PDGF, ИЛ-1, ИЛ-2 активно участвуют в мобилизации клеток воспаления, межклеточной кооперации) имеет перспективы, однако до сих пор не разработана. Ее можно будет проводить в определенные периоды жизни человека. Необходима также разработка средств, усиливающих выведение ХС.
Список использованной литературы
- Горев Н.Н., Кожура И.М., Костюк Л.В. и др. (1972) Экспериментальный атеросклероз и возраст. Медицина, Москва, 208 с.
- Грацианский Н.А. (2002) Очередное (окончательное?) подтверждение неэффективности антиоксидантных витаминов в профилактике коронарной болезни сердца и ее осложнений. Кардиология, 2: 1.
- Денисов Л.Н. (1997) Гиперхолестеринемия. В кн.: Ревматические болезни / Под ред. spy phone mobile В.А. Насоновой, Н.В. Бунчука, с. 471–474.
- Камышников В.С. (2004) Справочник по клинико-биохимическим исследованиям и лабораторной диагностике. МЕДпрессинформ, Москва, с. 511–526.
- Кольман Я., Рем К.Г. (2000) Наглядная биохимия: Пер. с немец. Мир, Москва, 469 с.
- Лопухин Ю.М. (1986) Холестериноз. Наука, Москва.
- Марри Р., Греннер Д., Мейес П. и др. (2004) Биохимия человека: Пер. с англ. Мир, Москва, 2: 414 с.
- Николаев А.Я. (2007) Биологическая химия. Медицинское информационное агентство, Москва, 568 с.
- Пауков В.С., Кауфман О.Я. (1995) Стадии воспаления. Формы воспаления. В кн.: Воспаление / Под ред. В.В. Серова, В.С. Паукова. Медицина, Москва, с. 176–199.
- Пауков В.С., Серов В.В. (1995) Сущность воспаления, его место в биологии и медицине. В кн.: Воспаление / Под ред. В.В. Серова, В.С. Паукова. Медицина, Москва, с. 30–35.
- Пальцев М.А., Аничков Н.М. (2001) Патологическая анатомия в 2 т. Медицина, Москва. Т. 2, с. 16.
- Саркисов Д.С., Серов В.В. (1995) История учения о воспалении. В кн.: Воспаление / Под ред. В.В. Серова, В.С. Паукова. Медицина, Москва, 640 с.
- Серов В.В., Пауков В.С. (ред.) (1995) Воспаление. Медицина, Москва, 640 с.
- Серов В.В., Шехтер А.Б. (1981) Соединительная ткань. Медицина, Москва, 312 с.
- Серов В.В. (1998) Воспаление. В кн.: Патологическая анатомия / Под ред. В.В. Серова, М.А. Пальцева. Медицина, Москва, с.146.
- Струков А.И., Кауфман О.Я. (1989) Гранулематозное воспаление и гранулематозные болезни. Медицина, Москва, 179 с.
- Струков А.И., Серов В.В., Каримов Д.С. (ред.) (1990) Общая патология человека. Руководство для врачей. Т. 1., Медицина, Москва, 448 с.
- Томпсон Г.Р. (1991) Руководство по гиперлипидемии: Пер. с англ. Лондон, 255 с.
- Шехтер А.Б., Серов В.В. (1995) Воспаление и регенерация. В кн.: Воспаление / Под ред. В.В. Серова, В.С. Паукова. Медицина, Москва, 640 с.
- Goldstein I., Brown M. (1977) Atherosclerosis The low-density lipoprotein receptor hypothesis. Metabolism, 26: 12–57.
- Epstein W.L. (1980) Foreing body granulomas. In: Basic and clinical aspects granulomatous diseases. Ed. Boros D.L., Yochida T. — New York, р. 133–148.
- Fuster V. (1994) Mechanisms leading to myocardial infarction: insights from stadies of vascular biology. Circulation, 90: 2126–2146.
- Libby P. (1995) Molecular bases of the coronary syndromes. Circulation, 91: 2844–2851.
- Ross R. (1999) Atherosclerosis — an inflammatory disease. N. Engl. J. Med., 340: 115–126.
Адрес для переписки:
Казимирко Виталий Казимирович
04112, Киев, ул. Дорогожицкая, 9
Национальная медицинская академия
последипломного образования
имени П.Л. Шупика
Leave a comment